Patent application title: Retroviral vectors
Inventors:
Alan John Kingsman (Oxford, GB)
Susan Mary Kingsman (Oxford, GB)
Narry Kim (Seoul, KR)
Kyriacos Mitrophanous (Oxford, GB)
IPC8 Class: AC12N1500FI
USPC Class:
4353201
Class name: Chemistry: molecular biology and microbiology vector, per se (e.g., plasmid, hybrid plasmid, cosmid, viral vector, bacteriophage vector, etc.) bacteriophage vector, etc.)
Publication date: 2009-11-26
Patent application number: 20090291491
Inventors list |
Agents list |
Assignees list |
List by place |
Classification tree browser |
Top 100 Inventors |
Top 100 Agents |
Top 100 Assignees |
Usenet FAQ Index |
Documents |
Other FAQs |
Patent application title: Retroviral vectors
Inventors:
Kyriacos Mitrophanous
Susan Mary Kingsman
Alan John Kingsman
Narry Kim
Agents:
FROMMER LAWRENCE & HAUG
Assignees:
Origin: NEW YORK, NY US
IPC8 Class: AC12N1500FI
USPC Class:
4353201
Patent application number: 20090291491
Abstract:
Retroviral vector production systems for producing lentivirus-based vector
particles which are capable of infecting and transducing non-dividing
target cells, wherein one or more of the auxiliary genes such as vpr,
vif, tat, and nef in the case of HIV-1 are absent from the system. The
systems and resulting retrovirus vector particles have improved safety
over existing systems and vectors.Claims:
1-38. (canceled)
39. A retroviral vector capable of infecting and transducing a cell, wherein the retroviral vector is lentivirus-based and wherein one or more auxiliary genes chosen from the HIV-1 auxiliary genes vpr; vif, tat and nef or from the analogous auxiliary genes of other lentivirus, which auxiliary genes are normally present in the lentivirus on which the retroviral vector is based, is or are absent from the retroviral vector.
Description:
[0001]This is a continuation of U.S. application Ser. No. 11/646,041,
filed Dec. 27, 2006, which is a continuation of U.S. application Ser. No.
10/661,761, filed Sep. 11, 2003, now U.S. Pat. No. 7,198,784, which is a
continuation-in-part of U.S. application Ser. No. 09/915,169, filed Jul.
25, 2001, now U.S. Pat. No. 6,669,936, which is a divisional application
of U.S. application Ser. No. 09/224,014, filed Dec. 28, 1998, now U.S.
Pat. No. 6,312,682, which is a continuing application of PCT/GB97/02857,
filed Oct. 17, 1997 and claiming priority to Great Britain Patent
application Nos. 9621680.9, filed Oct. 17, 1996, and 9624457.9, filed
Nov. 25, 1996. This application makes reference to U.S. application Ser.
No. 10/408,456, filed on Apr. 7, 2003. All of the foregoing applications,
as well as all documents cited in the foregoing applications
("application documents") and all documents cited or referenced in the
application documents are incorporated herein by reference. Also, all
documents cited in this application ("herein-cited documents") and all
documents cited or referenced in herein-cited documents are incorporated
herein by reference. In addition, any manufacturer's instructions or
catalogues for any products cited or mentioned in each of the application
documents or herein-cited documents are incorporated by reference.
Documents incorporated by reference into this text or any teachings
therein can be used in the practice of this invention. Documents
incorporated by reference into this text are not admitted to be prior
art.
[0002]This invention relates to retroviral vector production systems and to retroviral vector particles produced by the systems. In particular, it relates to systems and vector particles from which certain retroviral auxiliary factors are absent. The invention also relates to uses of retroviral vectors, in particular for gene therapy.
[0003]Retroviral vectors have been the vehicle of choice for clinical gene transfer because of their efficacy, safety, and stable long-term gene expression. According to the United States National Institutes of Health RAC report issued in September 1996 (Ross et al., 1996), 76 out of 107 trials reviewed by the NIH were based on vector systems derived from the murine leukaemia virus (MLV).
[0004]One major drawback of these vectors is their inability to infect non-proliferating cells such as neurons, macrophages and haematopoietic stem cells. These cells are important targets for gene therapy.
[0005]Human immunodeficiency virus type 1 (HIV-1) belongs to a sub-family within the retroviruses, the lentiviruses and in common with other members of this family HIV can infect quiescent cells. This makes lentiviruses attractive vectors for gene therapy.
[0006]The viral determinants for HIV-1 infection of non-dividing cells are thought to reside in the p17 matrix protein (MA) and vpr (Gallay et al., 1996). MA has karyophilic properties conferred by a conserved stretch of basic residues, which constitute a nuclear localization signal (NLS) (Bukrinsky et al., 1993). Vpr also contains a distinct NLS (Mahalingam et al., 1995). MA-NLS mutant viruses fail to replicate efficiently in macrophages in the absence of a functional vpr gene (Heinzinger et al., 1994). These data have been interpreted to mean that vpr as well as MA function as karyophilic determinants of HIV-1. In the absence of vpr the transduction efficiency of monocyte-derived macrophages decreases by over 50%, in the presence of functional MA. (Naldini et al., 1996).
[0007]Following work reported in Lever et al., 1989 which showed the sequences required for packaging of HIV-1, there has been much interest in the development of an HIV-1 based gene therapy vector. Transfer of foreign genes into a human T-cell line by a replication defective HIV-1 based vector was demonstrated by Poznanski et al. (Poznansky et al., 1991). Other groups have designed HIV-1 based vectors that are tat-inducible (Buchschacher, Jr. and Panganiban, 1992) or that use heterologous promoters (Shimada et al., 1991). However, the viral titers obtained with these vectors was low (at most 103 infectious particles per ml), and it was not clear whether the vector system could guarantee the production of helper virus-free vectors. More recently, new efforts to produce helper virus-free vectors have been based on three-plasmid cotransfections (Richardson et al., 1995). HIV vectors can be pseudotyped with Vesicular Stomatitis Virus glycoprotein (VSV-G) and these particles retain infectivity after concentration by ultracentrifugation (Akkina et al., 1996). Pseudotyping with VSV-G confers a broader host range and eliminates the chances of recombination to produce wild type HIV envelope. In vivo transduction of non-dividing neuronal cells has been demonstrated with VSV-G pseudotyping of HIV-1 in a three-plasmid cotransfection system (Naldini et al., 1996 and Naldini et al., 1996a).
[0008]HIV-1 contains nine genes, three of which: gag, pol and env are found in all retroviruses. These are the structural genes. The other six: vif, vpu, vpr, nef, tat and rev are referred to as auxiliary genes. Other retroviruses have different sets of auxiliary genes in their wild type genomes. Some of the auxiliary genes of other retroviruses are analogous to those of HIV-1, although they may not always have been given the same names in the literature. Analogous auxiliary genes have homology in their nucleotide sequences and perform the same or similar functions. HIV-2 and SIV strains generally contain env, vpr, vif, tat, and nef genes analogous to those of HIV-1. HIV-2 and some strains of SIV also contain vpx which, in some SIV strains lacking vpr can be considered analogous to vpr. Lentiviruses other than HIV-1 also contain auxiliary genes which are not analogous to the HIV-1 auxiliary genes. Retrovirus auxiliary genes are reviewed for example by Tomonaga and Mikami (1996) and by Joag et al. in Fields Virology, Vol 2.
[0009]To date all vector systems based on HIV contain some or all of the HIV auxiliary genes. Rev acts as an RNA export protein and tat is a major transactivator of the proviral long terminal repeat (LTR). The auxiliary genes play a crucial role in viral replication and pathogenesis. The auxiliary genes have not been fully characterized nor their function defined.
[0010]However some of the auxiliary genes are thought to be involved in the pathogenesis of HIV-1. Tat has been implicated in the development of Kaposi's sarcoma (Barillari et al., 1993; Ensoli et al., 1990). HIV vpr has been shown to cause cell arrest and apoptosis and this has been proposed to be the cause of T-Cell dysfunction seen in AIDS patients (Jowett et al., 1995). Also extracellular Vpr present in peripheral blood has been suggested to contribute to tissue-specific pathologies associated with HIV infection since Vpr induces cell proliferation and differentiation (Levy et al., 1993 and Levy et al., 1995).
[0011]Since the roles of the auxiliary genes are not clear and they probably play a major role in pathogenesis their removal from HIV-1 vector production systems is desirable, provided that sufficiently high retrovirus vector titer and ability to transduce non-proliferating cells can be retained.
[0012]Naldini et al's data shows that the presence or absence of vpu has no effect on the vector particle titer. That is, a packaging system they used produced a titer of 4×105 when pseudotyped with VSV-G and this system was env and vpu negative. In another system which was only env negative they obtained the same titer (Naldini et al., 1996 and Naldini et al., 1996a). However, as already discussed another system of Naldini et al. which was vpr negative as well as vpu negative gave a transduction efficiency which was decreased by 50% compared to a vpr positive system.
[0013]We have now discovered that leaving some or all of the auxiliary genes out of retrovirus vector production systems does not significantly compromise vector particle titers or the ability of the vector particles to transduce non-dividing cells.
[0014]The invention therefore provides in one aspect a retroviral vector production system for producing lentivirus-based, replication defective vector particles for gene therapy, said vector particles capable of infecting and transducing non-dividing mammalian target cells, which system comprises a set of nucleic acid sequences encoding the components of the vector, wherein one or more functional genes chosen from the HIV-1 auxiliary genes vpr, vif, tat and nef or from the analogous auxiliary genes of other lentiviruses, which auxiliary genes are normally present in the lentivirus on which the vector particles are based, is or are absent from the system. The functional vpu gene may also be absent, with the proviso that when the production system is for an HIV-1 based vector and vpr and vpu are both absent, so also is one of the other auxiliary genes.
[0015]In another aspect, the invention provides retroviral vector particles produced by a retroviral vector particle production system described herein.
[0016]In yet another aspect, the invention provides a DNA construct for use in a retroviral vector production system described herein, said DNA construct encoding a packagable RNA vector genome for a retroviral vector particle and operably linked to a promoter, wherein all of the functional retroviral auxiliary genes are absent from the construct, other than rev which is optionally present. The DNA construct may be provided as part of a set of DNA constructs also encoding some or all of the structural components of the vector particles.
[0017]In further aspects, the invention provides the use of retroviral vector particles as described herein, for gene therapy and in the preparation of a medicament for gene therapy; and a method of performing gene therapy on a target cell which method comprises infecting and transducing the target cell using a retroviral vector particle as described herein. The invention further provides transduced target cells resulting from these uses and methods. The invention thus provides a gene delivery system for use in medicine.
[0018]The expression "lentivirus-based" means that the vector particles are derived from a lentivirus. The genome of the vector particle comprises components from the lentivirus as a backbone. The vector particle as a whole contains essential vector components compatible with the RNA genome, including reverse transcription and integration systems. Usually these will include the gag and pol proteins derived from the lentivirus.
[0019]Being derived from a lentivirus, the retroviral vector particles are capable of infecting and transducing non-dividing cells. Thus, the vector particles are able to deliver a selected gene or genes such as therapeutically active genes, to the genome of a target cell. During the infection process, lentiviruses form a pre-integration complex in the target cell cytoplasm containing integrase, core proteins and proviral DNA. The complex is able to pass across the nuclear membrane of the target cell, by means of signal sequences in the proteins. Non-lentiviral retroviruses either lack the proteins or have the proteins but without the appropriate signal sequences.
[0020]Examples of lentiviruses are HIV-1 and HIV-2, SIV, FIV, BLV, EIAV, CEV and visna virus. Of these, HIV and SIV are presently best understood. However, a non-immunodeficiency virus may be preferred for use in gene therapy because the immunodeficiency viruses inevitably bring with them safety considerations and prejudices.
[0021]The absence of functional auxiliary genes from the retroviral vector production system means that those functional genes will also be absent from retroviral vector particles produced by the system. Also, any auxiliary proteins that would otherwise be encoded by those genes and incorporated into the vector particles, will be absent from the vector particles. In known retroviral vector production systems, the auxiliary genes may be present as part of the vector genome-encoding DNA, or together with the packaging components. The location of an auxiliary gene in a vector production system depends in part on its relationship with other retroviral components. For example, vif is often part of a gag-pol packaging cassette in a packaging cell. Thus, to remove a functional auxiliary gene for the purposes of the invention may involve its removal from the packaging components, or from the vector genome, or perhaps both.
[0022]To remove a functional auxiliary gene may not require removal of the gene in its entirety. Usually removal of part of the gene, or disruption of the gene in some other way will be sufficient. The absence of a functional auxiliary gene is understood herein to mean that the gene is not present in a form in which it is capable of encoding the functional auxiliary protein.
[0023]In a preferred system according to the invention, functional vpr and tat genes or analogous genes normally present in the lentivirus on which the vector particles are based are both absent. These two auxiliary genes are associated with characteristics of lentiviruses which are particularly undesirable for a gene therapy vector. However, other than by the proviso given above, the invention is not limited with regard to the combination of auxiliary genes that are absent. In a system according to the invention for producing HIV-1-based vector particles, any combination of three, or more preferably four, of the genes may be absent in their functional form. Most preferably, all five of the auxiliary genes vpr, vif, tat, nef, and vpu are absent in their functional form. Similarly, for systems concerned with other lentiviruses, it is most preferable that all of the auxiliary genes are absent in their functional form (except rev which is preferably present unless replaced by a system analogous to the rev/RRE system).
[0024]In order to ensure efficient export of RNA transcripts of the vector genome from the nucleus to the cytoplasm, it is preferable to include functional rev and rev response element (RRE) sequences in the vector genome, or to include alternative sequences in the genome which perform the same function as the rev/RRE system. For example, a functional analogue of the rev/RRE system is found in Mason Pfizer monkey virus. This is known as CTE and consists of an RRE-type sequence in the genome which is believed to interact with a factor in the infected cell. The cellular factor can be thought of as a rev analogue. Thus, CTE may be used as an alternative to the rev/RRE system.
[0025]As will be evident, in order to function as a vector the retroviral vector particles described herein will need to have a reverse transcription system (compatible reverse transcription and primer binding sites) and an integration system (compatible integrase and integration sites) allowing conversion to the provirus and integration of the double-stranded DNA into the target cell genome. Additionally, the vector genome will need to contain a packaging signal. These systems and signals will generally be derived from the lentivirus on which the vector is based. It will be evident that although the vector according to the invention is based on a lentivirus, the elements of the lentivirus incorporated into the vector may be genetically or otherwise altered versions of the elements in the wild type lentivirus. Alterations may be achieved by manipulating either the RNA genome or other components of the retroviral vector particle production system. For example, portions of the lentivirus genome not required for the vector can be excluded. Also, the vector production system can employ substitutes e.g. for the lentivirus env gene, to give the vector a different target cell range (this is known as pseudotyping).
[0026]A retroviral vector particle according to the invention carries one or more selected genes for delivery to a target cell. The selected genes are chosen according to the effect sought to be achieved. For gene therapy purposes there will be at least one therapeutically active gene encoding a gene product which is active against the condition it is desired to treat or prevent. Additionally there may be a selected gene which acts as a marker by encoding a detectable product. Therapeutic genes may encode for example an antisense RNA, a ribozyme, a transdominant negative mutant of a target protein, a toxin, a conditional toxin, an antigen that induces antibodies or helper T-cells or cytotoxic T-cells, a single chain antibody or a tumour suppressor protein.
[0027]Preferably the construction of the vector genome is such that in the DNA provirus, the therapeutic gene or genes is or are under transcriptional control of the 5' LTR but not otherwise operably linked to any other promoter from the vector. Thus, expression of the gene or genes is in a single transcription unit. Preferably also the 5' LTR is a modified lentivirus LTR for which the promoter function is not tat-dependent. This may be achieved by replacing the R and U3 lentivirus promoter functions by alternative promoter functions, which may be derived from another retrovirus or may be of non-retroviral origin. A strategy for this is described in Cannon et al. 1996 and in the Examples.
[0028]It will be evident that the term "gene" is used loosely here, and includes any nucleic acid coding for the desired polypeptide or RNA. Usually, genes delivered by vectors according to the invention will be cDNAs.
[0029]Retroviral vector particles according to the invention will also be capable of infecting and transducing cells which are slowly-dividing, and which non-lentiviruses such as MLV would not be able to efficiently infect and transduce. Slowly-dividing cells divide once in about every three to four days. Mammalian non-dividing and slowly-dividing cells include brain cells, stem cells, terminally differentiated macrophages, lung epithelial cells and various other cell types. Also included are certain tumour cells. Although tumours contain rapidly dividing cells, some tumour cells especially those in the centre of the tumour, divide infrequently.
[0030]The DNA construct encoding the vector genome described wherein is preferably linked to a high efficiency promoter such as the CMV promoter. Other high efficiency promoters are known. This gives rise to a high level of expression of the vector RNA by the retroviral vector production system.
[0031]Suitable host or producer cells for use in the retroviral vector production system according to the invention are well known in the art. Many retroviruses have already been split into replication defective genomes and packaging components. For those which have not the technology is available for doing so. The producer cell encodes the viral components not encoded by the vector genome such as the gag, pol and env proteins. The gag, pol and env genes may be introduced into the producer cell and stably integrated into the cell genome to give a packaging cell line. The retroviral vector genome is then introduced into the packaging cell line by transfection or transduction to create a stable cell line that has all of the DNA sequences required to produce a retroviral vector particle. Another approach is to introduce the different DNA sequences that are required to produce a retroviral vector particle e.g. the env coding sequence, the gag-pol coding sequence and the defective retroviral genome into the cell simultaneously by transient triple transfection. In a preferred system according to the invention, both the structural components and the vector genome will all be encoded by DNA stably integrated into a host cell genome.
[0032]Accordingly, it is an object of the invention to not encompass within the invention any previously known product, process of making the product, or method of using the product such that Applicants reserve the right and hereby disclose a disclaimer of any previously known product, process, or method. It is further noted that the invention does not intend to encompass within the scope of the invention any product, process, or making of the product or method of using the product, which does not meet the written description and enablement requirements of the USPTO (35 U.S.C. 112, first paragraph) or the EPO (Article 83 of the EPC), such that Applicants reserve the right and hereby disclose a disclaimer of any previously described product, process of making the product, or method of using the product.
[0033]It is noted that in this disclosure and particularly in the claims and/or paragraphs, terms such as "comprises", "comprised", "comprising" and the like can have the meaning attributed to it in U.S. patent law; e.g., they can mean "includes", "included", "including", and the like; and that terms such as "consisting essentially of" and "consists essentially of" have the meaning ascribed to them in U.S. patent law, e.g., they allow for elements not explicitly recited, but exclude elements that are found in the prior art or that affect a basic or novel characteristic of the invention.
[0034]These and other embodiments are disclosed or are obvious from and encompassed by, the following description.
[0035]The following description, given by way of example, but not intended to limit the invention solely to the specific embodiments described, may best be understood in conjunction with the accompanying drawings, in which:
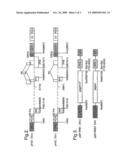
[0036]FIG. 1 shows a vector production system according to the invention, using a three-plasmid co-transfection of 293T cells;
[0037]FIG. 2 shows HIV-based vector genomes for use in the invention;
[0038]FIG. 3 shows HIV-1 gag-pol gene expression plasmids for use in the invention; and
[0039]FIG. 4 shows transduction efficiencies for vectors according to the invention lacking the five auxiliary factors.
[0040]To produce a safe HIV packaging system devoid of all unnecessary genes, we have developed a system which does not contain vpr, nef, tat, vif or vpu (FIG. 1.). The packaging components were placed on three separate plasmids and overlapping sequences were minimized ensuring no recombination and no helper virus production. This HIV vector has been shown to transduce aphidicolin treated non-dividing cells in the absence of vpr. Titers were obtained that are similar to the Naldini et al. titers for systems which contain all the auxiliary genes (Naldini et al., 1996a).
[0041]This is the first minimal lentiviral vector system. The fact that high titers are observed with this system shows that the auxiliary genes (except rev) are redundant for the production of high titers and for the transduction of non-dividing cells. This is contrary to the assumption made by Naldini et al. that the reason for the production of high titer virus stocks is due to the incorporation of accessory proteins (such as nef) into the viral particle (Naldini et al., 1996).
[0042]The system may have additional advantages for HIV therapy. Replacing the HIV-1 LTR with a different promoter such as a constitutive HCMV promoter permits the use of anti-Tat molecules such as Tat transdominant mutants (Echetebu et al., 1994) or TAR decoys (Lisziewicz et al., 1993) as therapeutic agents as they will not affect vector production.
[0043]It will be evident that minimal lentiviral vectors as described herein, lacking all of the wild-type virus auxiliary genes, may also have applications as vaccines.
[0044]The invention will now be further described by way of the following non-limiting examples which further illustrate the invention, and are not intended, nor should they be interpreted to, limit the scope of the invention.
EXAMPLES
Materials and Methods
Plasmid Construction
[0045]pGP-RRE1 is a pWl3 (Kim et al., 1989) derived gagpol vif expression plasmid. The RRE of pWl3 (Accession number: U26942) was inserted by blunt-ending the Sty I/Sty I fragment (7720-8050) into pBluescript KS+ Sma I cut creating pBSRRE. The Nar I/Eco RI fragment of pWl3 (637-5743) was inserted into pBSRRE cut with Cla I and Eco RI to create pBSGPRRE1. The Xho I/Not I fragment (containing gagpol and RRE) was inserted into the expression plasmid pCl-Neo to create pGR-RRE1. To remove the vif coding region, pBSGPRRE1 was cut with NdeI and SmaI, blunt-ended and was relegated to generate pBSGPRRE2. The gagpol gene and RRE were inserted into pCl-neo in XhoI and NotI site to make pGP-RRE2.
[0046]The construction of pTIN406, pTIN408 and pTIN414 has been described (Cannon et al., 1996). The 5' LTR of pH3Z and pH4Z contain a CMV promoter at the U3 position and the HIV R and U5 regions. HIVdge was made from HIVgpt (Page et al., 1990) by blunt-ending the Cla I site (829) to create a frameshift mutation. HIVdge was cut with Bgl II and Pst I (473-1414) and inserted into pTIN406. pTIN406 has an LTR structure of CMV, R (HIV) and U5 (MLV). This created a hybrid LTR containing CMV, and R, U5 from HIV called pBS5'. To provide the plasmid with rev and RRE the Eco RI/Xho I fragment (5743-8897) was cut from HIVdge1.2 which is a HIVdge derivative containing a deletion from Nde I to Bgl II (6403-7621) and was inserted into pBS5' to create pBS5'R. The 3' LTR was provided by inserting the Not I/Xho I fragment of pBS3' into pBS5'R creating pH2. pBS3' was created by a three way ligation of the Xho I/Hind III fragment of pWl3, the Hind III/Kpn I fragment of pTIN408 into pBluescript KS+ (Xho I/Kpn I). A CMV promoter was inserted into the unique Xho I site of pH2 from pSPCMV (Sal I/Xho 1) making pH2CMV. pSPCMV was created by inserting pLNCX (Accession number: M28246) (Pst I/Hind III) into pSP72 (Promega). The β-galactosidase gene was inserted from PTIN414 into pSP72 (Xho I/Sph I) to make pSPlacZ. A Xho I/Sal I digest of pSPlacZ gave the β-galactosidase coding region which was inserted into pH2-CMV to give pH3Z. pH4Z was constructed to create tat-deficient vector. The first 50 bp of the tat-coding region was removed by replacing EcoRI (5743)I-SpeI fragment in pH3 with EcoRI (5881)-SpeI PCR product amplified using PCR primers DELT5 (5'-CGTGAATTCGCCTAAAACTGCTTGTACCA-3') (SEQ ID NO: 1) and DELT3 (5'-GAACTAATGACCCCGTAATTG-3') (SEQ ID NO: 2) to create pH4. The Nsi I/Spe I fragment from pH4 was inserted into pH3Z to generate pH4Z.
[0047]A vpr expression plasmid was constructed by PCR amplification of the vpr coding region from pNL4.3 (Accession number: U26942) using the following primers: 5' primer GCGAATTCGGATCCACCATGGAACAAGCCCCAGAAGAC (5563-5583) (SEQ ID NO: 3) and 3' primer GCGAATTCGGATCCTCTAGGATCTACTGGC TCCATT (5834-5853) (SEQ ID NO: 4). This amplicon was cloned into pLIGATOR (R & D Systems). The expression plasmid pCl-vpr was made by inserting the Mlu I and Xho I fragment containing the vpr coding region into pCl-Neo (Promega).
[0048]pAC29.1 was cut by Bam HI to give the VSV-G coding region which was inserted into pSA91 (Bgl II).
Cell Lines
[0049]293T (293ts/A1609) (DuBridge et al., 1987) cells were maintained in Dulbelco's modified Eagle's medium (GIBCO), HeLa cells and 208F cells in MEM (GIBCO), all of which containing 10% (v/v) fetal calf serum supplemented with antibiotics.
Production and Assays of Vectors
[0050]Retroviral vector stocks were produced according to our previously published protocol (Soneoka et al., 1995). Briefly, human kidney 293T (1.5×106) cells were plated on 10-cm plates and transiently transfected with 15 mg of each plasmid (gag-pol and env expression plasmids together with a vector plasmid) by calcium phosphate DNA precipitation (Chen and Okayama, 1987). The culture supernatants were harvested 36 hours later, filtered through 0.45 mm and either used immediately or frozen at -70° C. Transduction was carried out by adding virus onto target cells for 2 hours, in the presence of 8 mg/ml polybrene followed by the addition of fresh media. 5-bromo-4-chloro-3-indolyl b-D-galactoside (X-Gal) was used to measure the expression of β-galactosidase 48 hours later, as previously described (Soneoka et al., 1995). Titers were obtained by counting the number of lac z (blue foci) forming units per ml (l.f.u./ml). G1/S phase arrested cultures were prepared by adding aphidicolin (5 mg/ml) 24 hours before infection and then daily throughout the experiment.
Results
HIV Vector Production
[0051]H3Z (tat positive) and H4Z (tat negative) are HIV-1 based vectors designed to be produced by three plasmid co-transfection into 293T cells (FIG. 2). For efficient packaging by the HIV cores, the vectors contain the first 778 bases of gag but a frameshift mutation, introduced 40 bp from the ATG start codon, prevents the expression of gag proteins. RRE was included to boost packaging efficiency and rev is expressed from the vector to support the HIV mRNA export. The internal CMV promoter-driven β-galactosidase gene was inserted to serve as a reporter gene. For both the vector genomes transcription is driven by a CMV promoter which has been used to replace the 5' LTR U3. This makes the vector genome tat independent. Two HIV-1 gagpol constructs were made (FIG. 3); pGP-RRE1 (vif positive) and pGP-RRE2 (vif negative). Since the gagpol genes have been inserted into pCl-neo which is a CMV driven expression plasmid gagpol expression is tat independent. pRV67, the VSV glycoprotein construct was used for the pseudotyping. By placing the different genes on different plasmids the probability of generating replication competent virus by recombination could be minimized.
Transduction Efficiency of the Vector
[0052]Replication defective retroviral particles were generated by transient co-transfection of human kidney 293T cells with the three plasmids described above and either used immediately or frozen at -70° C. The different vector constructs were used to produce virus. It was found that the minimal constructs (H4Z and pGP-RRE2) gave comparable titers to that of the vif, vpr, nef and tat positive viruses (Table 1).
[0053]When the minimal system was tested on various cell lines the titers differed (Table 2). The vectors yielded titers of 3.2×105 l.f.u./ml with polybrene treatment, 9.1×104 l.f.u./ml without polybrene treatment in 293T cells. Also the same vectors, without polybrene, yielded 9.6×103 l.f.u./ml and 8.3×103 l.f.u./ml in HeLa and 208F cells, respectively. These titers are comparable with those obtained by Naldini et al., 1996 (Naldini et al., 1996), which are the highest ones published so far.
Effect of vpr on the Transduction of Aphidicolin-Treated Cells
[0054]To test the effect of vpr on non-dividing cell transduction, vpr was included in the packaging system by co-transfection of pCl-vpr along with pH4Z, pGP-RRE2 and pRV67 plasmids. The transduction efficiencies of the viral particles generated were assayed on growing and growth-arrested 293T cells and HeLa cells (FIG. 4). MLV-derived packaging and transducing vectors (Soneoka, 1995) served as controls. HeLa cells and 293T cells were growth-arrested at G1/S phase by aphidicolin treatment. The minimal HIV vector H4Z was as efficient at transducing G1/S-arrested as proliferating HeLa and 293T cells, whereas the MLV-based vector was only 0.002% as effective.
[0055]Vpr-deficient H4Z could transduce the growth-arrested cells as efficiently as vpr-containing vector, suggesting that HIV-1 MA is sufficient for providing the vector with the ability to transduce non-dividing cells.
CONCLUSION
[0056]We have set up an HIV-1 based vector production system, which does not contain vpr, vpu, nef, vif and tat based on a three-plasmid co-transfection method. This vector can transduce proliferating cells with a titer of up to 3.2×105 l.f.u./ml, which is comparable to other MLV-based vectors and can easily be increased by concentration using ultracentrifugation (data not shown). No helper virus has been detected (data not shown).
[0057]This minimal vector has been demonstrated to transduce growth-arrested HeLa cells and 293T cells as efficiently as vpr, vif, nef and tat containing vectors. Therefore it can be concluded that only rev is required for the production of high titer HIV based vectors and that these vectors can transduce non-dividing cells.
[0058]This is the first report of the construction of a high titer minimal lentiviral vector that can transduce non-dividing cells. The removal of five out of the six auxiliary genes (except rev) and the minimal sequence overlap between the plasmids makes this system the safest one to date for the production of HIV-vectors for gene therapy.
Figure Legends
[0059]FIG. 2. HIV vector genomes. The numbers indicate the coordinates from HXB2. HCMV promoter (-597 to -1). HIV sequences (455 to 1415; 5743 (H3Z) or 5881 (H4Z) to 6403; 7621 to 8897; 8897 to 9720) from HXB2. HCMV promoter as an internal promoter (900 bp). Cloning site (XhoI). Backbone; pBluescriptKS+.
[0060]FIG. 3. HIV-1 gag-pol gene expression plasmids. HIV-1 gagpol coding region and RRE was cloned into pCl-neo (PROMEGA) at XhoI and NotI site.
[0061]FIG. 4. Transduction of non-dividing cells. Transduction efficiencies of the H4Z vectors were measured by X-gal staining and are shown in Y-axis as l.f.u./ml. G1/S phase arrested cells were prepared by treating the cells with aphidicolin (5 μg/ml).
REFERENCES
[0062]Akkina, R. K., Walton, R. M., Chen, M. L., Li, Q. X., Planelles, V., and Chen, I. S. (1996). J. Virol. 70, 2581-2585. [0063]Barillari, G., Gendelman, R., Gallo, R. C., and Ensoli, B. (1993). Proc. Natl. Acad. Sci. U.S.A. 90, 7941-7945. [0064]Buchschacher, G. L., Jr. and Panganiban, A. T. (1992). J. Virol. 66, 2731-2739. [0065]Bukrinsky, M. I., Haggerty, S., Dempsey, M. P., Sharova, N., Adzhubel, A., Spitz, L., Lewis, P., Goldfarb, D., Emerman, M., and Stevenson, M. (1993). Nature 365, 666-669. [0066]Cannon, P. M., Kim, N., Kingsman, S. M., and Kingsman, A. J. (1996). J. Virol. 70, 8234-8240. [0067]Chen, C. and Okayama, H. (1987). Mol. Cell Biol. 7, 2745-2752. [0068]Echetebu, C. O., H. Rhim, C. H. Herrmann and A. P. Rice (1994), J. Acquired Immune Defic. Syndrome. 7, 655-664. [0069]Ensoli, B., Barillari, G., Salahuddin, S. Z., Gallo, R. C., and Wong Staal, F. (1990). Nature 345, 84-86. [0070]Gallay, P., Stitt, V., Mundy, C., Oettinger, M., and Trono, D. (1996). J. Virol. 70, 1027-1032. [0071]Heinzinger, N. K., Bukinsky, M. I., Haggerty, S. A., Ragland, A. M., Kewalramani, V., Lee, M. A., Gendelman, H. E., Ratner, L., Stevenson, M., and Emerman, M. (1994). Proc. Natl. Acad. Sci. U.S.A. 91, 7311-7315. [0072]Joag, S. V., Stephens, E. B. and Narayan, O. in Fields Virology, Vol 2, 1970-1982 (Lippincott-Raven Publishers). [0073]Jowett, J. B., Planelles, V., Poon, B., Shah, N. P., Chen, M. L., and Chen, I. S. (1995). J. Virol. 69, 6304-6313. [0074]Kim, S. Y., Byrn, R., Groopman, J., and Baltimore, D. (1989). J. Virol. 63, 3708-3713. [0075]Lever, A., Gottlinger, H., Haseltine, W., and Sodroski, J. (1989). J. Virol. 63, 4085-4087. [0076]Levy, D. N., L. S. Fernandes, W. V. Williams, and D. B. Weiner (1993), Cell, 72, 541-50. [0077]Levy, D. N., Y. Refae; and D. B. Weiner (1995), J. Virol., 69, 1243-52. Lisziewicz, J., D. Sun, J. Smythe, P. Lusso, F. Lori, A. Louie, P. Markham, J. Rossi, M. Reitz and R. C. Gallo (1993), Proc. Natl. Acad. Sci, USA, 90, 8000-4. [0078]Mahalingam, S., Collman, R. G., Patel, M., Monken, C. E., and Srinivasan, A. (1995). Virology 212, 331-339. [0079]Naldini, L., Blomer, U., Gallay, P., Ory, D., Mulligan, R., Gage, F. H., Verma, I. M., and Trono, D. (1996). Science 272, 263-267. [0080]Naldini, L., Blomer, U., Gage, F. H., Trono, D., and Verma, I. M. (1996). Proc. Natl. Acad. Sci. U.S.A. 93, 11382-11388. [0081]Page, K. A., Landau, N. R., and Littman, D. R. (1990). J. Virol. 64, 5270-5276. Poznansky, M., Lever, A., Bergeron, L., Haseltine, W., and Sodroski, J. (1991). J. Virol. 65, 532-536. [0082]Richardson, J. H., Kaye, J. F., Child, L. A., and Lever, A. M. (1995). J. Gen. Virol. 76, 691-696. [0083]Ross, G., Erickson, R., Knorr, D., Motulsky, A. G., Parkman, R., Samulski, J., Straus, S. E., and Smith, B. R. (1996). Hum. Gene Ther. 7, 1781-1790. Shimada, T., Fujii, H., Mitsuya, H., and Nienhuis, A. W. (1991). J. Clin. Invest. 88, 1043-1047. [0084]Tomonaga, K. and Mikami, T. (1996). J. General Virol. 77, 1611-1621.
[0085]All publications mentioned in the above specification are herein incorporated by reference. Various modifications and variations of the described methods and system of the invention will be apparent to those skilled in the art without departing from the scope and spirit of the invention. Although the invention has been described in connection with specific preferred embodiments, it should be understood that the invention as claimed should not be unduly limited to such specific embodiments. Indeed, various modifications of the described modes for carrying out the invention which are apparent to those skilled in molecular biology or related fields are intended to be within the scope of the following claims.
TABLE-US-00001 TABLE 1 Effects of accessory gene expression on vector titer. Accessory genes Plasmids Titer Tat Vif Nef Vpr Vector Gagpol Nef Vpr (I.f.u./ml)a + + - + pH3Z pGP-RRE1 pCI- 2.2 × 105 Vpr + + + - pH3Z pGP-RRE1 pC- 2.5 × 105 Nef + + - - pH3Z pGP-RRE1 4.0 × 105 + - - - pH3Z pGP-RRE2 3.7 × 105 - - - - pH4Z pGP-RRE2 4.6 × 105 aTransduction efficiency was measured in 293T cells by counting the number of blue colonies following X-gal staining 48 hours after transduction and were indicated as lacZ colony forming unit per ml virus stock (I.f.u./ml).
TABLE-US-00002 TABLE 2 Transduction efficiency of the minimal H4Z vector on various cell lines. Titer (l.f.u./ml)a Cell line Without polybrene With polybrene 293T Human kidney 9.1 × 104 3.2 × 105 HeLa Human epithelium 9.6 × 103 N.D. 208F Rat fibroblast 8.3 × 103 N.D. aTransduction efficiency was measured by counting the number of blue colonies following X-gal staining 48 hours after transduction and were indicated as lacZ colony forming unit per ml virus stock (l.f.u./ml).
Sequence CWU
1
4129DNAArtificial Sequenceoligonucleotide 1cgtgaattcg cctaaaactg cttgtacca
29221DNAArtificial
Sequenceoligonucleotide 2gaactaatga ccccgtaatt g
21338DNAArtificial Sequenceoligonucleotide
3gcgaattcgg atccaccatg gaacaagccc cagaagac
38436DNAArtificial Sequenceoligonucleotide 4gcgaattcgg atcctctagg
atctactggc tccatt 36
User Contributions:
comments("1"); ?> comment_form("1"); ?>Inventors list |
Agents list |
Assignees list |
List by place |
Classification tree browser |
Top 100 Inventors |
Top 100 Agents |
Top 100 Assignees |
Usenet FAQ Index |
Documents |
Other FAQs |
User Contributions:
Comment about this patent or add new information about this topic:
| People who visited this patent also read: | |
| Patent application number | Title |
|---|---|
| 20120148602 | Cancer Treatment With Endothelin Receptor Antagonists |
| 20120148601 | BACTERIAL SUPERANTIGEN VACCINES |
| 20120148600 | Anti-CD79B Antibodies and Immunoconjugates and Methods of Use |
| 20120148599 | METHODS AND COMPOSITIONS FOR NEURAL DISEASE IMMUNOTHERAPY |
| 20120148598 | METHODS FOR TREATING A TUMOR USING AN ANTIBODY THAT SPECIFICALLY BINDS GRP94 |
Images included with this patent application:
 |  |
 |  |
 |  |
| Similar patent applications: | |
| Date | Title |
|---|---|
| 2010-07-22 | Targeting pseudotyped retroviral vectors |
| 2010-12-23 | Polypurine tract modified retroviral vectors |
| 2008-11-20 | Retroviral vector |
| 2010-04-29 | Method of adenoviral vector synthesis |
| 2010-08-19 | Non-replicating paramyxoviridae virus vector |
| New patent applications in this class: | |
| Date | Title |
|---|---|
| 2019-05-16 | Selective recovery |
| 2019-05-16 | Methods and compositions for attenuating anti-viral transfer vector igm responses |
| 2019-05-16 | Immunomodulatory oncolytic adenoviral vectors, and methods of production and use thereof for treatment of cancer |
| 2017-08-17 | Novel nucleic acid vector |
| 2017-08-17 | Devices and methods for plasmid purification |
| New patent applications from these inventors: | |
| Date | Title |
|---|---|
| 2017-06-22 | Virus purification |
| 2016-11-17 | Viral vector production system |
| 2013-09-12 | Antigen |
| 2013-05-09 | Catecholamine enzyme fusions |
| 2011-03-03 | Antibodies |
| Top Inventors for class "Chemistry: molecular biology and microbiology" | |
| Rank | Inventor's name |
|---|---|
| 1 | Marshall Medoff |
| 2 | Anthony P. Burgard |
| 3 | Mark J. Burk |
| 4 | Robin E. Osterhout |
| 5 | Rangarajan Sampath |