Patent application title: PROKARYOTIC DNA REPAIR LIGASES
Inventors:
Aidan Doherty (Cambridge, GB)
Marina Della (Cambridge, GB)
Geoffrey Weller (Cambridge, GB)
Stephen Jackson (Cambridge, GB)
IPC8 Class: AC12P1934FI
USPC Class:
435 9152
Class name: Polynucleotide (e.g., nucleic acid, oligonucleotide, etc.) acellular preparation of polynucleotide involving a ligase (6.)
Publication date: 2011-12-29
Patent application number: 20110318789
Abstract:
The present invention relates to the cloning and characterisation of a
prokaryotic DNA repair ligase, which is shown to possess a range of
activities that allow the ligation and repair of non-compatible DNA ends
and double strand breaks (DSBs). The enzyme has a range of applications
in the manipulation and cloning of nucleic acids.Claims:
1. A method of modifying a nucleic acid molecule comprising; contacting
the nucleic acid molecule with a prokaryotic DNA repair ligase
polypeptide.
2. A method according to claim 1 wherein the prokaryotic DNA repair ligase polypeptide comprises one or more of: a primase domain, a nuclease domain, and a ligase domain, said one or more domains sharing greater than 20% sequence identity with the corresponding domain sequence of Mt-Lig (CAB08492).
3. A method according to claim 1 wherein the prokaryotic DNA repair ligase polypeptide shares greater than 20% sequence identity with the sequence of Mt-Lig (CAB08492).
4. A method according to claim 1 wherein the prokaryotic DNA repair ligase polypeptide is Mt-Lig (CAB08492) or a variant or allele thereof.
5. A method according to claim 1 wherein the nucleic acid molecule and the Mt-Lig polypeptide are contacted in the presence of a prokaryotic Ku polypeptide.
6. A method according to claim 5 wherein the prokaryotic Ku polypeptide shares greater than 20% sequence identity with the sequence of Mt-Ku (CAB08491).
7. A method according to claim 6 wherein the prokaryotic Ku polypeptide is Mt-Ku (CAB08491) or an allele or variant thereof.
8. A method of ligating nucleic acid molecule ends comprising; contacting a first nucleic acid end and a second nucleic acid end with an prokaryotic DNA repair ligase polypeptide, wherein said first and said second nucleic acid ends are non-compatible.
9. A method according to claim 8 wherein said first and said second nucleic acid ends comprise non-complementary overhang regions.
10. A method according to claim 8 wherein the first end is on a first nucleic acid molecule and the second end is on a second nucleic acid molecule.
11. A method according to claim 10 wherein the first and second nucleic acid molecules are DNA.
12. A method according to claim 10 wherein the first nucleic acid molecule is DNA and the second nucleic acid molecule is RNA.
13. A method according to claim 8 wherein the first and second ends are on the same nucleic acid molecule.
14. A method according to claim 8 comprising isolating and/or purifying the ligated nucleic acid molecule.
15. A method of labelling a nucleic acid molecule comprising; contacting a nucleic molecule having a first terminus with an prokaryotic DNA repair ligase polypeptide in the presence of labelled nucleotides.
16. A method according to claim 15 wherein the nucleotides are NTPs.
17. A method according to claim 15 wherein the nucleotides are dNTPs.
18. A method of filling in a single stranded gap in a double stranded nucleic acid molecule comprising; contacting a double stranded nucleic acid molecule having a single stranded region with an prokaryotic DNA repair ligase polypeptide.
19. A method according to claim 18 wherein said nucleic acid molecule and said prokaryotic DNA repair ligase polypeptide are contacted in the presence of NTPs.
20. A method according to claim 18 wherein said nucleic acid molecule and said prokaryotic DNA repair ligase polypeptide are contacted in the presence of dNTPs.
21. A method of removing a single stranded overhang from the end of a nucleic acid molecule comprising; contacting said nucleic acid molecule with a prokaryotic DNA repair ligase polypeptide
22. A method according to claim 21 wherein the prokaryotic DNA repair ligase polypeptide is an Mt-Lig polypeptide.
23. A method according to claim 21 wherein said nucleic acid molecule is contacted in the presence of Mg2+ or Mn2+.
24. A method of producing an RNA molecule comprising; contacting a prokaryotic DNA repair ligase polypeptide and a template DNA strand in the presence of NTPs.
25. A method according to claim 24 wherein prokaryotic DNA repair ligase and template DNA are contacted in the presence of a primer oligonucleotide.
26. A method of producing a DNA molecule comprising; contacting A prokaryotic DNA repair ligase polypeptide and a nucleic acid template in the presence of dNTPs and a primer oligonucleotide.
27. A method according to claim 26 wherein the nucleic acid template is an RNA template.
28. A method according to claim 26 wherein the nucleic acid template is an DNA template.
29. A method according to claim 8 wherein the prokaryotic DNA repair ligase polypeptide comprises one or more of: a primase domain, a nuclease domain, and a ligase domain, said one or more domains sharing greater than 20% sequence identity with the corresponding domain sequence of Mt-Lig (CAB08492).
30. A method according to claim 8 wherein the prokaryotic DNA repair ligase polypeptide shares greater than 20% sequence identity with the sequence of Mt-Lig (CAB08492).
31. A method according to claim 8 wherein the prokaryotic DNA repair ligase polypeptide is Mt-Lig (CAB08492) or a variant or allele thereof.
32. A method according to claim 8 wherein the nucleic acid molecule and the Mt-Lig polypeptide are contacted in the presence of a prokaryotic Ku polypeptide.
33. A method according to claim 32 wherein the prokaryotic Ku polypeptide shares greater than 20% sequence identity with the sequence of Mt-Ku (CAB08491).
34. A method according to claim 32 wherein the prokaryotic Ku polypeptide is Mt-Ku (CAB08491) or an allele or variant thereof.
35. A kit comprising an isolated Mt-Lig polypeptide for use in a method according to claim 1.
36. A kit according to claim 35 comprising an isolated Mt-Ku polypeptide.
37. A kit according to claim 35 comprising dNTPs.
38. A kit according to claim 35 comprising NTPs.
39. A kit according to claim 35 comprising one or more of buffers, stabilisers and excipients.
40. A method of producing a prokaryotic DNA repair polypeptide comprising; (a) causing expression from nucleic acid which encodes a prokaryotic DNA repair polypeptide in a suitable expression system to produce the polypeptide recombinantly; and, testing the recombinantly produced polypeptide for prokaryotic DNA repair activity.
41. A method according to claim 40 wherein the recombinantly produced polypeptide is tested for one or more of: non-complementary end ligation activity, DNA dependent RNA primase activity, 3'-5' exonuclease activity, DNA and RNA dependent DNA polymerase activity, DNA dependent RNA polymerase activity, ATP dependent DNA and RNA ligase activity and DNA terminal transferase activity.
42. A method according to claim 39 wherein the prokaryotic DNA repair polypeptide is an Mt-Lig polypeptide or an allele or variant thereof.
43. A method according to claim 40 comprising purifying said recombinantly produced polypeptide.
Description:
CROSS-REFERENCE TO RELATED APPLICATIONS
[0001] This is a continuation of co-pending U.S. application Ser. No. 10/568,055, filed Sep. 27, 2006, which is the §371 U.S. national stage of International Application No. PCT/GB2004/003349, filed Aug. 2, 2004, which claims the benefit of U.S. Provisional Patent Application No. 60/494,088, filed Aug. 12, 2003. All of the above-listed applications are incorporated herein in their entirety.
[0002] This invention relates to methods and reagents for the manipulation and modification of nucleic acid molecules.
[0003] Double-strand breaks (DSBs) in DNA arise during exposure to ionizing radiation (IR) and as intermediates during site-specific rearrangement events such as mating-type switching in Saccharomyces cerevisiae and V(D)J recombination in vertebrates (Critchlow and Jackson (1998) Trends Biochem Sci 23 394). In eukaryotic cells, the primary DNA end-binding component of non-homologous end-joining (NHEJ), Ku, is a heterodimer of two sequence-related subunits (Ku70: 69 kD and Ku80: 83 kD) (Gell & Jackson (1999) Nucl Acid Res 17 3494) that forms an open ring-like structure through which a variety of DNA end structures can be threaded (Walker et al (2001) Nature 412 607). DNA-bound Ku helps to recruit the ligase IV/XRCC4 complex, thereby enhancing its ligation activity (McElhinny et al (2000) Mol. Cell. Biol. 20 2996). In vertebrates, Ku also recruits the DNA-dependent protein kinase catalytic subunit (DNA-PKcs), thereby activating its kinase activity, which is required for DSB rejoining (Dvir et al. (1992) PNAS USA 89 11920). Mammalian cells deficient in these NHEJ proteins are defective in DSB rejoining and are hypersensitive to IR (Smith & Jackson (1999) Genes Dev 13 916).
[0004] In contrast to the conservation between these components in higher and lower eukaryotes, NHEJ has not been reported in prokaryotes, although genes with homology to Ku70 and Ku80 have been identified in some bacterial genomes (Doherty et al (2001) FEBS Lett 500 186; Aravind & Koonin (2001) Genome Res 11 1365).
[0005] The present inventors have identified and characterised a prokaryotic polypeptide that is involved in NHEJ and has a range of enzymatic activities relating to the modification of nucleic acid molecules. These activities are useful in the manipulation of nucleic acid in a range of molecular biology applications.
[0006] An aspect of the invention provides a method of modifying a nucleic acid molecule comprising; [0007] contacting the nucleic acid molecule with a prokaryotic DNA repair ligase polypeptide.
[0008] A prokaryotic DNA repair ligase polypeptide may comprise an amino acid sequence from a prokaryotic cell which shares greater than about 20% sequence identity with the sequence of Mt-Lig (CAB08492; SEQ ID NO: 91), greater than about 30%, greater than about 40%, greater than about 50%, greater than about 60%, greater than about 70%, greater than about 80%, greater than about 90% or greater than about 95% with the given amino acid sequence.
[0009] A prokaryotic ligase may comprise one or more of: a primase domain, a nuclease domain, and a ligase domain. In some embodiments, a prokaryotic ligase may comprise all three domains.
[0010] A primase domain may share greater than about 20% sequence identity with the sequence of Mt-Lig (CAB08492; SEQ ID NO: 91) between residues 1-324, greater than about 30%, greater than about 40%, greater than about 50%, greater than about 60%, greater than about 70%, greater than about 80%, greater than about 90% or greater than about 95% with the given amino acid sequence.
[0011] A nuclease domain may share greater than about 20% sequence identity with the sequence of Mt-Lig (CAB08492; SEQ ID NO: 91) between residues 325-447, greater than about 30%, greater than about 40%, greater than about 50%, greater than about 60%, greater than about 70%, greater than about 80%, greater than about 90% or greater than about 95% with the given amino acid sequence.
[0012] A ligase domain may share greater than about 20% sequence identity with the sequence of Mt-Lig (CAB08492; SEQ ID NO: 91) between residues 448-759, greater than about 30%, greater than about 40%, greater than about 50%, greater than about 60%, greater than about 70%, greater than about 80%, greater than about 90% or greater than about 95% with the given amino acid sequence.
[0013] In some embodiments, a prokaryotic DNA repair ligase polypeptide may comprise one or more conserved motifs as shown in FIG. 4 and/or table 2.
[0014] Suitable prokaryotic DNA repair ligase polypeptides may include an Mt-lig polypeptide as described below, a B. subtilis YkoU polypeptide, a Bacillus halodurans BH2209 polypeptide, a Pseudomonas aeruginosa PA2150 polypeptide, a Archaeoglobus fulgidus AF11725 polypeptide, Mesorhizobium loti Mll2077, Mll4606, Mll9625 polypeptides, Sinorhizobium loti SMB20685, SMA0424 polypeptides, Agrobacterium tumefaciens AGR_L--502P and AGR_PAT--68 polypeptides or variants or alleles of these polypeptides.
[0015] In some preferred embodiments, the prokaryotic DNA repair ligase polypeptide is an Mt-lig polypeptide. An Mt-lig polypeptide may comprise or consist of the amino acid sequence of database accession number CAB08492 which is encoded by the M. tuberculosis ORF RV0938 (Z95209) or may be a variant or allele of this sequence.
[0016] A gene encoding a prokaryotic DNA repair ligase may be functionally linked with a gene encoding a prokaryotic Ku polypeptide, for example within an operon of the prokaryotic genome.
[0017] In some embodiments, a substrate nucleic acid molecule may be contacted with a prokaryotic DNA repair ligase polypeptide in the presence of a prokaryotic Ku polypeptide.
[0018] A prokaryotic Ku polypeptide may comprise an amino acid sequence from a prokaryotic cell which shares greater than about 20% sequence identity with the sequence of Mt-Ku (CAB08491; SEQ ID NO: 92), greater than about 30%, greater than about 40%, greater than about 50%, greater than about 60%, greater than about 70%, greater than about 80%, greater than about 90% or greater than about 95% with the given amino acid sequence.
[0019] Suitable prokaryotic Ku polypeptides may include Mt-Ku, B. subtilis YkoV, M. Loti Mlr9623, Mlr9624, B. halodurans BH2209 and A fulgidus AF172, or variants or alleles thereof.
[0020] In preferred embodiments, the prokaryotic Ku polypeptide is an Mt--Ku polypeptide. An Mt--Ku polypeptide may comprise or consist of the amino acid sequence of database accession number CAB08491 that is encoded by the M. tuberculosis ORF RV0937c (Z95209) or may be a variant or allele of this sequence.
[0021] The production of suitable prokaryotic DNA repair ligases and prokaryotic Ku polypeptides is described in more detail below.
[0022] An allele or variant may have an amino acid sequence which differs from a given sequence, by one or more of addition, substitution, deletion and insertion of one or more amino acids but which still has substantially the same sequence as the given sequence. Such an addition, substitution, deletion or insertion may represent a natural variation which occurs between individuals within a species and which has no phenotypic effect. An allele or variant may comprise one or more conserved motifs as shown in FIG. 4 and/or table 2.
[0023] A polypeptide which is an amino acid sequence variant or allele may comprise an amino acid sequence which differs from a given amino acid sequence, but which shares greater than about 50% sequence identity with such a sequence, greater than about 60%, greater than about 70%, greater than about 80%, greater than about 90% or greater than about 95%. A variant or allelic sequence may share greater than about 60% similarity, greater than about 70% similarity, greater than about 80% similarity or greater than about 90% similarity with a given amino acid sequence.
[0024] Amino acid similarity and identity are generally defined with reference to the algorithm GAP (GCG Wisconsin Package®, Accelrys, San Diego Calif.). GAP uses the Needleman & Wunsch algorithm to align two complete sequences that maximizes the number of matches and minimizes the number of gaps. Generally, the default parameters are used, with a gap creation penalty=12 and gap extension penalty=4. Use of GAP may be preferred but other algorithms may be used, e.g. BLAST or TBLASTN (which use the method of Altschul et al. (1990) J. Mol. Biol. 215: 405-410), FASTA (which uses the method of Pearson and Lipman (1988) PNAS USA 85: 2444-2448), or the Smith-Waterman algorithm (Smith and Waterman (1981) J. Mol. Biol. 147: 195-197), generally employing default parameters.
[0025] Similarity allows for "conservative variation", i.e. substitution of one hydrophobic residue such as isoleucine, valine, leucine or methionine for another, or the substitution of one polar residue for another, such as arginine for lysine, glutamic for aspartic acid, or glutamine for asparagine.
[0026] Particular amino acid sequence alleles or variants may differ from that a given sequence by insertion, addition, substitution or deletion of 1 amino acid, 2, 3, 4, 5-10, 10-20, 20-30, or 30-50 amino acids
[0027] A polypeptide for use in a method of the invention may comprise a fragment of a sequence described herein, for example a fragment comprising a primase, nuclease or ligase domain.
[0028] A nucleic acid molecule for use in a method of the invention may be linear, with two ends or termini. The ends may independently be blunt-ended or comprise 3' or 5' overhangs.
[0029] The nucleic acid molecule may be wholly or partially synthetic and may include genomic DNA, cDNA, RNA or a fragment thereof.
[0030] In some preferred embodiments, the nucleic acid molecule is double-stranded. A double-stranded nucleic acid molecule may be modified, for example, by ligating an end of the molecule with an end of either the same or a different nucleic acid molecule, removing 3' overhangs at the ends and filling-in single stranded `gap` regions.
[0031] In other preferred embodiments, the nucleic acid molecule is single-stranded. A single-stranded nucleic acid molecule may be modified, for example, by acting as a template for DNA or RNA polymerase activity to generate a complementary strand
[0032] Certain preferred embodiments relate to the inter- or intra-molecular ligation of nucleic acid using prokaryotic DNA repair ligase polypeptides.
[0033] A method of ligating double-stranded nucleic acid ends may comprise; [0034] contacting a first nucleic acid end and a second nucleic acid end with a prokaryotic DNA repair ligase polypeptide, such as an Mt-ligase polypeptide.
[0035] The first and second nucleic acid ends may be the termini of double stranded nucleic acid molecules, for example, double stranded DNA molecules.
[0036] The first and second nucleic acid ends may be on the same nucleic acid molecule (i.e. an intramolecular ligation reaction) or may be on different nucleic acid molecules (i.e. a first and a second nucleic acid molecule joined in an intermolecular ligation reaction).
[0037] In some embodiments, one nucleic molecule joined by the prokaryotic DNA ligase may be DNA and the other nucleic acid molecule may be RNA.
[0038] For example, a method of joining double-stranded nucleic acid termini may comprise; [0039] contacting a first nucleic molecule having a first terminus and a second nucleic acid molecule having a second terminus with a prokaryotic DNA repair ligase polypeptide as described above, [0040] said first and second termini being joined by said polypeptide, [0041] wherein the first nucleic acid molecule is DNA and the second nucleic acid molecule is RNA.
[0042] In some embodiments, the ends or termini to be ligated are non-compatible. Non-compatible ends are non-complementary and therefore non-cohesive. Examples of non-compatible ends include ends created by enzymatic digestion with different restriction endonucleases (i.e. endonucleases which recognise different nucleotide target sequences). Non-compatible nucleic acid ends may comprise non-complementary single-stranded 5' or 3' overhang regions which do not naturally form base-pairs.
[0043] Nucleic acid ends may be contacted with a prokaryotic DNA repair ligase in the presence of a prokaryotic Ku polypeptide as described above. A suitable prokaryotic Ku polypeptide may comprise an amino acid sequence which is naturally associated with the prokaryotic DNA repair ligase, for example a prokaryotic Ku polypeptide from the same strain or species.
[0044] A nucleic acid molecule produced by ligation with a prokaryotic DNA repair ligase polypeptide described above may be isolated and/or purified and subjected to further manipulation using standard techniques.
[0045] A prokaryotic DNA repair ligase polypeptide, as described above, may also be useful in labelling nucleic molecules by means of a terminal transferase reaction.
[0046] A method of labelling a nucleic acid molecule may comprise; [0047] contacting a nucleic molecule having a first terminus with a prokaryotic DNA repair ligase polypeptide, such as an Mt-lig polypeptide, in the presence of labelled nucleotides.
[0048] Labelled nucleotides may be NTPs (i.e. GTP, ATP, TTP, UTP or CTP) or dNTPs (i.e. dGTP, dATP, dTTP, dUTP or dCTP).
[0049] A nucleotide may be labelled with a fluorophore such as FITC or rhodamine, a radioisotope, or a non-isotopic labeling reagent such as biotin or digoxigenin.
[0050] The DNA dependent RNA or DNA polymerase activity of Mt-lig polypeptide may be useful in filling in gaps (i.e. repairing single stranded regions) in a double stranded nucleic molecule.
[0051] A method of filling in a single stranded gap in a double stranded nucleic acid molecule may comprise; [0052] contacting a double stranded nucleic acid molecule having a single stranded region with a prokaryotic DNA repair ligase polypeptide, such as an Mt-lig polypeptide, in the presence of NTPs or dNTPs.
[0053] The nucleic acid molecule may be a DNA molecule and may be linear or circular.
[0054] NTPs or dNTPs may be used as substrates for the Mt-ligase polypeptide. A method may be used to fill in a gap in a dsDNA sequence with DNA or with a `patch` of RNA. This may be useful in a range of applications such as producing DNA substrates with defined labelled patches of DNA or RNA that could be used to study DNA repair, recombination and replication processes using these novel substrates both in vivo and in vivo.
[0055] The exonuclease activity of the prokaryotic DNA repair ligase polypeptide may also be useful in blunt ending double stranded nucleic acid and removing single-stranded overhangs.
[0056] A method of blunt-ending a nucleic acid molecule may comprise; [0057] contacting said nucleic acid molecule comprising a single stranded overhang with a prokaryotic DNA repair ligase polypeptide.
[0058] The nucleic acid molecule may contacted with the prokaryotic DNA repair ligase polypeptide in the presence of Mg2+ or Mn2+.
[0059] The overhang may be a 3' overhang.
[0060] A suitable prokaryotic DNA repair ligase polypeptide for use in blunt ending methods may comprise or consist of a prokaryotic DNA repair ligase nuclease domain as described above.
[0061] DNA dependent RNA polymerase activity of a prokaryotic DNA repair ligase polypeptide as described above may be used to produce RNA molecules.
[0062] A method of producing an RNA molecule may comprise; [0063] contacting a prokaryotic DNA repair ligase polypeptide, such as an Mt-lig polypeptide and a template DNA strand in the presence of NTPs.
[0064] Prokaryotic DNA repair ligase polypeptides are shown herein to possess an RNA primase activity which allows RNA to be synthesised without a primer sequence. In other embodiments, a primer may be desirable and the prokaryotic DNA repair ligase polypeptide and template DNA may be contacted in the presence of a primer oligonucleotide.
[0065] The RNA strand synthesised by the prokaryotic DNA repair ligase polypeptide may be isolated and/or purified, for example from the template DNA by reverse phase liquid chromatography or digestion with a DNA nuclease.
[0066] The DNA polymerase activity of a prokaryotic DNA repair ligase polypeptide may be used to produce a DNA molecule.
[0067] A method of producing an DNA molecule may comprise; [0068] contacting a prokaryotic DNA repair ligase polypeptide and a template nucleic acid strand in the presence of dNTPs and a primer oligonucleotide.
[0069] Prokaryotic DNA repair ligase polypeptides such as Mt-lig polypeptide are shown herein to possess a DNA dependent DNA polymerase activity and an RNA dependent DNA polymerase (i.e. reverse transcriptase). Suitable template nucleic acid strand may therefore be either DNA or RNA.
[0070] Other aspects of the invention relate to kits and reagents for use in molecular biology applications.
[0071] A composition for use in a method described above may comprise an isolated prokaryotic DNA repair ligase polypeptide, for example a Mt-lig polypeptide, and an isolated prokaryotic Ku polypeptide, such as Mt-Ku. The composition may further comprise buffers, stabilisers, excipients, Mg2+ and/or Mn2+. A composition may also comprise dNTPs or NTPs.
[0072] Reagents for use in a method as described herein, such as isolated prokaryotic DNA repair ligase polypeptide, may be provided as part of a kit, e.g. in a suitable container such as a vial in which the contents are protected from the external environment. In preferred embodiments, the kit also comprises an Mt-Ku polypeptide as described above. The kit may include instructions for use of the polypeptide e.g. in a method described above. A kit may include one or more other reagents required for the method, such as buffers, excipients, stabilisers, NTPS, dNTPs, labelled NTPs/dNTPs, Mg2+ or Mn2+. A kit may also include vessels such as tubes or cuvettes suitable for use in carrying out the method.
[0073] Another aspect of the invention provides a kit comprising an isolated prokaryotic DNA repair ligase polypeptide such as Mt-lig polypeptide and, optionally an isolated prokaryotic Ku polypeptide, such as Mt-Ku, for use in a method of modifying a nucleic acid molecule as described above.
[0074] Other aspects of the invention relate to the production of prokaryotic DNA repair ligase polypeptides such as Mt-lig polypeptide.
[0075] A method of producing a prokaryotic DNA repair ligase polypeptide may comprise;
(a) causing expression from nucleic acid which encodes a prokaryotic DNA repair ligase polypeptide in a suitable expression system to produce the polypeptide recombinantly; (b) testing the recombinantly produced polypeptide for prokaryotic DNA repair ligase polypeptide activity.
[0076] Prokaryotic DNA repair ligase polypeptide activity may include one or more of the following: non-complementary end ligation activity, DNA dependent RNA primase activity, 3'-5' exonuclease activity, DNA and RNA dependent DNA polymerase activity, DNA dependent RNA polymerase activity, ATP dependent DNA and RNA ligase activity and DNA terminal transferase activity.
[0077] Determination of one or more of these activities may be performed using standard techniques in the art (for example, see Sambrook & Russell, Molecular Cloning, A Laboratory Manual, Cold Spring Harbor Laboratory Press, 2001, and Ausubel et al, Short Protocols in Molecular Biology, John Wiley and Sons, 1992).
[0078] Suitable prokaryotic DNA repair ligase polypeptides are described above and include a B. subtilis YkoU polypeptide, a Bacillus halodurans BH2209 polypeptide, a Pseudomonas aeruginosa PA2150 polypeptide, a Archaeoglobus fulgidus AF11725 polypeptide, Mesorhizobium loti Mll2077, Mll4606, Mll9625 polypeptides, Sinorhizobium loti SMB20685, SMA0424 polypeptides, Agrobacterium tumefaciens AGR_L--502P and AGR_PAT--68 polypeptides, Mt-Lig and variants or alleles of these polypeptides.
[0079] Methods for the production of a recombinant polypeptide from encoding nucleic acid are well known in the art. Nucleic acid sequences encoding a Mt-lig polypeptide may be readily prepared by the skilled person using the information and references contained herein and techniques known in the art (for example, see Sambrook & Russell, Molecular Cloning, A Laboratory Manual, Cold Spring Harbor Laboratory Press, 2001, and Ausubel et al, Short Protocols in Molecular Biology, John Wiley and Sons, 1992), given the nucleic acid sequence and clones available. These techniques include (i) the use of the polymerase chain reaction (PCR) to amplify samples of such nucleic acid, e.g. from the M. tuberculosis genome, (ii) chemical synthesis, or (iii) preparing cDNA sequences. DNA encoding Mt-lig polypeptides may be generated and used in any suitable way known to those of skill in the art, including by taking encoding DNA, identifying suitable restriction enzyme recognition sites either side of the portion to be expressed, and cutting out said portion from the DNA. The portion may then be operably linked to a suitable promoter in a standard commercially available expression system. Another recombinant approach is to amplify the relevant portion of the DNA with suitable PCR primers.
[0080] In order to obtain expression of nucleic acid sequences, the sequences can be incorporated in a vector having one or more control sequences operably linked to the nucleic acid to control its expression. The vectors may include other sequences such as promoters or enhancers to drive the expression of the inserted nucleic acid, and/or nucleic acid sequences so that the polypeptide or peptide is produced as a fusion. Polypeptide can then be obtained by transforming the vectors into host cells in which the vector is functional, culturing the host cells so that the polypeptide is produced and recovering the polypeptide from the host cells or the surrounding medium. Prokaryotic cells are used for this purpose in the art, including strains of E. coli. The protein may also be expressed using the eukaryotic insect cell baculovirus expression system.
[0081] Suitable vectors can be chosen or constructed, containing appropriate regulatory sequences, including promoter sequences, terminator fragments, polyadenylation sequences, enhancer sequences, marker genes and other sequences as appropriate. Vectors may be plasmids, viral e.g. `phage, or phagemid, as appropriate. For further details see, for example, Molecular Cloning: a Laboratory Manual: 3rd edition, Sambrook et al. (2001) Cold Spring Harbor Laboratory Press. Many known techniques and protocols for manipulation of nucleic acid, for example in preparation of nucleic acid constructs, mutagenesis, sequencing, introduction of DNA into cells and gene expression, and analysis of proteins, are described in detail in Current Protocols in Molecular Biology, Ausubel et al. eds., John Wiley & Sons, 1992.
[0082] Following production, a polypeptide may be isolated and or purified using standard techniques.
[0083] Other aspects of the invention provide an isolated nucleic acid comprising a nucleotide sequence encoding a prokaryotic DNA repair ligase polypeptide as described above operably linked to a heterologous regulatory element, an expression vector comprising such a nucleic acid and a host cell, for example a prokaryotic host cell such as an E. coli cell, comprising such an expression vector.
[0084] An isolated nucleic acid comprising a nucleotide sequence encoding a prokaryotic DNA repair ligase polypeptide may further comprise a nucleotide sequence encoding a prokaryotic Ku polypeptide that is operably linked to a heterologous regulatory element.
[0085] Prokaryotic DNA repair ligase polypeptides, prokaryotic Ku polypeptides and encoding nucleic acids are described in more detail above.
[0086] Regulatory elements, expression vectors and host cells suitable for the expression of an Mt-lig polypeptide or other prokaryotic DNA repair ligase polypeptide are well-known in the art.
[0087] Various further aspects and embodiments of the present invention will be apparent to those skilled in the art in view of the present disclosure. All documents mentioned in this specification are incorporated herein by reference in their entirety.
[0088] The invention encompasses each and every combination and sub-combination of the features that are described above.
[0089] Certain aspects and embodiments of the invention will now be illustrated by way of example and with reference to the figures described below.
[0090] FIG. 1 shows the arrangement of the DNA ligase and Ku genes in the Ku-like gene operon in of various prokaryotes.
[0091] FIG. 2 shows the domain structure of a variety of prokaryotic DNA repair ligases.
[0092] FIG. 3 shows a putative mechanism for Mt-Lig and Mt-Ku.
[0093] FIG. 4 shows the Mt-Lig gene with the principle catalytic domains indicated (primase domain 1-324, nuclease domain 325-447 and ligase domain 448-759). I represents conserved motif: RLVFDLDPGE (SEQ ID NO: 1), II represents SGSKGLHLYT (SEQ ID NO: 2) and III represents KVFVDW (SEQ ID NO: 3). Variants of motif I include RLVFDLDPGE (SEQ ID NO: 72); ELVFDIDMTD (SEQ ID NO: 75); and ELVFDIDMDD (SEQ ID NO: 81). Variants of motif II include SGSKGLHLYT (SEQ ID NO: 73); SGRRGVHCWV (SEQ ID NO: 76); SGRRGIHCWV (SEQ ID NO: 79); SGRRGAHCWV (SEQ ID NO: 82); and SGRRGIHAWI (SEQ ID NO: 84). Variants of motif III include KVFVDW (SEQ ID NO: 74); FPRLDI (SEQ ID NO: 77); FPRLDV (SEQ ID NO: 78); YPRLDI (SEQ ID NO: 80); YPKLDV (SEQ ID NO: 83); and YPRLDV (SEQ ID NO: 85).
[0094] FIG. 5 shows constructs used to assay the activities of Mt-Ligase in the experiments described herein. FIG. 5(A) left panel shows a DNA duplex that forms a non-ligatable one nucleotide gap which is efficiently filled by Mt-Lig. FIG. 5(A) right panel shows a DNA duplex having a phosphate group added to the 5' terminus at the gap.
[0095] FIG. 5(B) shows a DNA duplex construct with a 3'-overhang.
[0096] FIG. 5(C) shows a DNA duplex construct containing non-ligatable one nucleotide gaps and a single stranded flap region.
[0097] FIG. 6 shows constructs used in assays for joining of DNA molecules with incompatible ends by Mt NHEJ.
[0098] FIG. 6(A) and FIG. 6(B) show DNA duplexes for assaying Mt-Lig activity.
[0099] FIG. 6 (C) shows a schematic of a plasmid repair assay, as described herein.
[0100] FIG. 7 shows a schematic of the interaction of the nuclease, polymerase and ligase activities of Mt-Lig in NHEJ. The sequences shown are CTGCAGATCATGCGCCGGATTGCCCC (SEQ ID NO: 20); GGTACGTGGTTTC (SEQ ID NO: 86); CGGCGCATGATCTGCAG (SEQ ID NO: 87); GAAACCACGTACCGGGGTGT (SEQ ID NO: 88); CTGCAGATCATGCGCCGGATTGCCCCGGTACGTGGTTTC (SEQ ID NO: 89); and GAAACCACGTACCGGGGCAATCCGGCGCATGATCTGCAG (SEQ ID NO: 90).
[0101] FIG. 8 shows the frequencies of gene conversion and simple religation NHEJ in wild-type and yku70 mutant yeast demonstrating reconstitution of NHEJ by combined expression of Mt-Ku and Mt-Lig.
[0102] FIG. 9 shows combinations of yeast and Mt Ku and ligase genes tested for NHEJ function in the absence of the gene conversion donor. Labels indicate those functions that were present in the cell. For example, "yeast Lig" indicates the strain genotype yku70 DNL4, while "bacteria Ku" indicates the presence of only the Mt-Ku expression plasmid.
[0103] FIG. 10 shows that NHEJ catalyzed by Mt proteins in yeast is only partially dependent on an intact MRX complex. No Ade.sup.+ colonies were recovered from dn14 rad50 yeast with vectors only and so this combination is not plotted.
[0104] FIG. 11 shows the extent of +2 frame-shifted NHEJ determined as a fraction of the total NHEJ events. Mt NHEJ led to a markedly lower +2 frequency than did yeast NHEJ, even in wild-type yeast. No Ade.sup.+ colonies were recovered from yku70 yeast with vectors only and so this strain is not plotted.
[0105] FIG. 12 shows diagrams of the inferred NHEJ intermediates for the HO(+2) and HO(-1) events, the overhang-to-overhang NHEJ events that will give a +2 reading frame.
[0106] FIG. 13 shows schematics of the suicide deletion systems used herein.
[0107] FIG. 13(A) shows a system in which galactose induction leads to I-SceI-mediated cleavage of its gene cassette from chromosome XV. Repair of the resulting DSB by precise religation NHEJ leads to in-frame expression of the ADE2 reporter gene. Imprecise NHEJ or, when present, gene conversion with a frame-shifted ade2 fragment on chromosome V leads to an out-of-frame ade2 gene on chromosome XV.
[0108] FIG. 13(B) shows a similar system to that of FIG. 13(A), except using the HO endonuclease and no gene conversion donor. Also, the initial reading frame has been adjusted so that precise simple-religation NHEJ (i.e. a frame-shift of 0 relative to an intact HO cut site) yields an out-of-frame ade2 product, while imprecise NHEJ events that result in a +2 frame-shift (or equivalent) yield an in-frame ADE2 product.
[0109] Table 1 shows sequences of plasmids from plasmid rescue assays, which were transformed into bacteria and subsequently sequenced. The starting ends, final products, and inferred alignment intermediates are shown.
[0110] Table 2 shows the conserved regions of prokaryotic ligases.
EXAMPLES
Materials and Methods
Cloning of Rv0937c and Rv09380RFs.
[0111] Full-length sequences for M. tuberculosis Rv0937c and Rv0938 were amplified by PCR from H37Rv genomic DNA using the following primers:
[0112] Rv0937c (M. tuberculosis Ku, 274 amino acids, 30.9 kD) was amplified using 5' primer (5'-ATG CGA GCC ATT TGG ACG GG-3') (SEQ ID NO: 4) and 3' primer (5'-GGA TCC TCA CGG AGG CGT TGG GAC G-3') (SEQ ID NO: 5).
[0113] Rv0938 (M. tuberculosis ligase, 759 amino acids, 83.6 kD) was amplified using 5' primer (5'-ATG GGT TCG GCG TCG GAG CA-3') (SEQ ID NO: 6) and 3' primer (5'-TCC TCA TTC GCG CAC CAC CTC ACT GG-3') (SEQ ID NO: 7).
[0114] The 5' primers contained an Nde I site, and the 3' primers contained a Bam HI site. PCR products were cloned into pET16b (Novagen). All DNAs cloned from PCR products were sequenced to confirm that no mutations were introduced during PCR. Proteins over-expressed from this vector carry an extra 21 amino acids (2.5 kD) at the NH2-terminus of the protein, due to addition of a 10-His tag and a Factor Xa cleavage site.
Overexpression of RV0937c and RV0938.
[0115] Recombinant protein was produced by first transforming E. coli B834 (DE3) pLysS cells (Novagen) with the pET16b plasmid (containing either Rv0938 or Rv0937c) and then selecting a single colony which was grown overnight at 37° C. in 5 ml LB broth supplemented with ampicillin at 100 μg/ml and chloramphenicol at 34 μg/ml. The overnight culture was used to inoculate 1 liter of LB broth supplemented with ampicillin and chloramphenicol as before. This culture was grown at 37° C. until an OD600 of 0.6 was achieved. At this point the culture was removed from the incubator and cooled to room temperature in a water bath and IPTG was added to a final concentration of 0.5 μM, to induce the production of the recombinant protein.
[0116] The culture was then returned to the incubator and grown overnight at 28° C. The cells were pelleted for 20 min at 4000 g.
Purification of Mt-Ku (RV0937c)
[0117] After sonication, the cell supernatant was treated with 60% of a saturated ammonium sulfate solution, incubating on ice for 1 hour. This was spun down, and the pellet was carefully resuspended in buffer A (50 mM Tris pH 7.5, 60 mM NaCl, 30 mM imidazole, 17 μg/ml PMSF, 34 μg/ml benzamidine). The resuspended was then loaded onto a nickel agarose (Qiagen) column, washed with 60 mM imidazole, and the protein eluted with 300 mM imidazole. The 300-mM peak was then loaded onto a DEAE Sepharose fast flow column. The Ku protein eluted between 200 and 300 mM NaCl.
Purification of Mt-Lig (RV0938)
[0118] After sonication, the cell debris was removed by centrifugation. The supernatant pellet was then loaded onto a nickel agarose column (Qiagen), washed with 60 mM imidazole, and the protein eluted with 300 mM imidazole. The 300 mM peak was then loaded onto a 5 ml Hi-Trap Q-Sepharose column (Amersham Biosciences). The ligase eluted at around 300 mM NaCl, which corresponded to a single protein band at approximately 83 kD, the predicted size for the full length Rv0938 gene product.
Double-Stranded Ligation Assay
[0119] Equimolar concentrations of Mt-Lig, Ligase IV/XRCC4 or T4 DNA ligase were incubated for 2 hours in 30 μl reaction mixture (50 mM Triethanolamine, pH 7.5, 2 mM Mg(OAc)2, 2 mM DTT, 0.1 mg/ml BSA) or 1× reaction buffer for T4 DNA ligase (Roche) with 70 fmol of DNA ([γ-32P]ATP labelled on the 5' end). Double-stranded DNA fragments were produced from the Bluescript plasmid (Stratagene) to give substrates of 53 bp, and 445 bp, and 2.56 kbp with 4 by overhangs at each end, and a 157-bp substrate with a 4-bp and a 2-bp overhang. These cohesive ends were not complementary to limit circularization. Bluescript was digested initially with the restriction enzymes Pst I and Afl III (NEB) to produce the 445-bp and 2.56-kbp DNA fragments. The large fragment produced by the first digestion was subjected to a second double digest with Kpn I and Pvu II (NEB) to produce 53 by and 157 by fragments.
[0120] After incubation, the reactions were deproteinized, phenol/chloroform extracted and precipitated with Pellet-Paint co-precipitant (Novagen). Aliquots of the reactions were run on 0.8% agarose gels. Dried gels were analyzed and quantified using a STORM Phosphorlmager (Molecular Dynamics). Reactions with Ku heterodimer were preincubated for 15 min on ice with indicated amounts of Ku heterodimer, and ligation reaction was started by adding the enzyme and transfer to 37° C.
DNA and RNA Extension Assays
[0121] Equal amounts of the labelled and unlabelled oligonucleotides were annealed by incubation at 70° C. for 10 min, 50° C. for 10 min, 40° C. for 10 min, 18° C. for 10 min, and then on ice for 5 min, to generate a linear duplex with the desired nucleotide gap using the following pairs of oligonucleotides; 5'-32P labelled 15-mer (5'-CTGCAGCTGATGCGC-3') (SEQ ID NO: 8) annealed to 20-mer (5'ATCCGGCGCATCAGCTGCAG-3') (SEQ ID NO: 9); 5'-32P labelled 15-mer (5'-CTGCAGCT-GATGCGC-3') (SEQ ID NO: 8) annealed to 25-mer (5'-AGTCGATCCTGCGCATCATCTGCAG-3') (SEQ ID NO: 10); 5'-32P labelled 15-mer (5'-CTGCAGCTGATGCGC-3') (SEQ ID NO: 8) annealed to 41-mer (5'-ACCCGGGGATCCGTACAGTCTATCCGGCGCATCAGCTGCAG-3') (SEQ ID NO: 11).
[0122] Alignment of the complementary single strands generates a non-ligatable nick in the unlabelled strand and a single-nucleotide gap in the labelled strand. A similar strategy was used to construct pairs of duplexes with single-strand extensions that, when aligned, give differently sized gaps with and without single-strand flaps.
[0123] The duplexes (100 nM) were incubated with Mt-ligase as indicated in reaction mixtures (10 μl) containing 50 mM potassium acetate, 20 mM Tris-acetate, 10 mM magnesium acetate, 1 mM dithiothreitol, pH 7.9 @ 25° C., 0.05 mM of each of the four dNTPs or the four NTPs. The reactions were supplemented with 100 μg/ml BSA and incubated at 37° C. for 30 min.
[0124] The reactions were stopped by the addition of gel loading buffer (95% (v/v) formamide, 0.09% (w/v) bromphenol blue, and 0.09% (w/v) xylene cyanol). After separation by denaturing gel electrophoresis, labelled DNA molecules in the dried gel were detected and quantitated by Phosphor-Imager analysis or x-ray exposure.
Ligation of Breaks Assay
[0125] Linear duplexes with complementary single-strand ends were constructed by annealing pairs of oligonucleotides. Alignment of the complementary single strands generates a ligatable nick in both the unlabelled and labelled strand and a single-nucleotide gap in the labelled strand. A similar strategy was used to construct pairs of duplexes with single-strand extensions that, when aligned, give differently sized gaps with and without single-strand flaps. Equal amounts of the labelled and unlabelled duplexes (100 nM) were incubated with Mt-ligase in 50 mM Tris-HCl, 10 mM MgCl2, 10 mM DTT, 1 mM ATP, 25 μg/ml BSA, (pH 7.5 @ 25° C.), 0.05 mM of each of the four dNTPs or the four NTPs. The reaction was incubated at 37° C. for 30 min. In assays to measure both DNA synthesis and ligation, the 5' termini of unlabelled oligonucleotides were phosphorylated.
Terminal Transferase Assay
[0126] Reaction mixtures (10 μl) containing 25 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 1 mM DTT, 100 μg/ml BSA, 100 nM 5'-32P labeled 50mer substrate (5'-GTA ACA AAG TTT GGA TTG CTA CTG ACC GCT CTC GTG CTC GTC GCT GCG TT-3') (SEQ ID NO: 12), 3 μg Mt-lig, and, as indicated, 50 μM ATP or 50 μM dATP. Reactions were incubated at 25° C. for 2 h and terminated by the addition of 1 μl loading buffer. After heat denaturation at 90° C. for 2 min, 4 μl of each reaction was loaded onto a 10% polyacrylamide-8M urea gel. After separation by electrophoresis, labelled products were detected by phosphor-imager analysis.
Primase Assay
[0127] Reaction mixtures (10 μl) contained 25 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 1 mM DTT, 100 μg/ml BSA, 0.25 μg of M13 mp19 (Invitrogen), 0.25 μCi [α-32P] ATP, various amounts of Mt-Lig, and, as indicated, 50 μM each of either GTP, CTP and UTP or 50 μM dNTPs. Reactions were incubated at 25° C. for 2 h and terminated by the addition of 1 μl loading buffer (95% formamide, 0.03% each bromophenol blue and xylene cyanol). After heat denaturation at 90° C. for 2 min, 4 μl of each reaction was loaded onto a 15% polyacrylamide-8M urea gel. After separation by electrophoresis, labelled products were detected by phosphor-imager analysis.
Coupled DNA Synthesis and Ligation
[0128] Linear duplexes with complementary single strand ends were constructed by annealing the following pairs of oligonucleotides; 5'-32P labelled 50-mer (5'-GTC TGT CTC ACT ATT AGA ACC CTT TAG AGT CAT GCG TCG CGA GGC AAC GC-3') (SEQ ID NO: 13) annealed to 43-mer (5'-GCC TCG CGA CGC ATG ACT CTA AAG GGT TCT AAT AGT GAG ACA G-3') (SEQ ID NO: 14); 41-mer (5'-GCG ACG AGC ACG AGA GCG GTC AGT AGC AAT CCA AAC TTT GT-3') (SEQ ID NO: 15) annealed to 50-mer (5'-GTA ACA AAG TTT GGA TTG CTA CTG ACC GCT CTC GTG CTC GTC GCT GCG TT-3') (SEQ ID NO: 16). Equal amounts of labelled and unlabeled duplexes (100 nM of each) were incubated with various amounts of Mt-Lig in reaction mixtures (10 μl) containing 25 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 50 μM each of dNTPs and 1 mM ATP at 25° C. for 2 h. Reactions were terminated by the addition of 1 μl loading buffer. After heat denaturation at 90° C. for 2 min, 4 μl of each reaction was loaded onto a 10% polyacrylamide-8M urea gel. After separation by electrophoresis, labelled products were detected by phosphor-imager analysis.
Nuclease Assay
[0129] Linear duplexes with complementary single-strand ends were constructed by annealing pairs of oligonucleotides; labeled 52-mer (5'-CTG TCT GTC TCA CTA TTA GAA CCC TTT AGA GTC ATG CGT CGC GAG GCA ACG C-3') (SEQ ID NO: 17) annealed to 43-mer; 41-mer annealed to 50-mer. 5'-32P labelled 20-mer (5'-GAAACCACGTACCGGCGTGT-3') (SEQ ID NO: 18) annealed to 13mer (5'-CTTTGGTCGATGG-3') (SEQ ID NO: 19); 26mer (5'-CTGCAGATCATGCGCCGGATTGCCCC-3') (SEQ ID NO: 20) annealed to 17-mer (5'-GACGTCTAGTACGCGGC-3) (SEQ ID NO: 21). Alignment of the complementary single strands generates a ligatable nick in both the unlabelled and labelled strand and a single-nucleotide gap in the labelled strand. A similar strategy was used to construct pairs of duplexes with single-strand extensions that, when aligned, give differently sized gaps with and without single-strand flaps. Equal amounts of the labelled and unlabelled duplexes (100 nM) were incubated with Mt-ligase in 50 mM potassium acetate, 20 mM Tris-acetate, 10 mM magnesium acetate, 1 mM dithiothreitol, pH 7.9 @ 25° C. The reactions were supplemented with 100 μg/ml BSA and incubated at 37° C. for 30 min. The reactions were stopped by the addition of gel loading buffer (95% (v/v) formamide, 0.09% (w/v) bromphenol blue, and 0.09% (w/v) xylene cyanol). After separation by denaturing gel electrophoresis, labelled DNA molecules in the dried gel were detected and quantitated by Phosphor-Imager analysis or x-ray exposure. In assays to measure both DNA synthesis and ligation, the 5' termini of unlabelled oligonucleotides were phosphorylated.
Plasmid Repair Assays
[0130] pUC18 plasmid was cut with restriction enzymes to give different non-complementary overhangs, producing a linearised duplex approximately 400-600 bp smaller than the uncut plasmid. SmaI and AatII were used to give a blunt end and a 3' overhang, HindIII and EcoRI were used to give non-complementary 5' overhangs, cut plasmid was purified using the Qiagen gel extraction kit. The plasmid was cut in such a way as to remove a 400-600 bp region from the plasmid. The reactions were carried out in 20 ul, with T4 ligase buffer (NEB), 50 uM dNTPs or NTPs, 50 nmol of cut plasmid, with Mt ligase (4 pmol) and Mt-Ku (0.05, 0.1, 0.5, or 1 pmol) as indicated. For controls, T4 ligase (0.2 units). The reactions were incubated with Mt ku for 20 minutes on ice before addition of Mt ligase, then the reactions were incubated at 37° C. for 1 hour.
[0131] PCR primers were produced to amplify across the region removed by restriction digest of the plasmid. The PCR reaction was carried out using Vent polymerase (NEB). Each reaction contained 100 pmol forward and reverse primers, Thermophil buffer (NEB), 2 mM dNTPs, 3 mM MgSO4, 1 ul Vent Polymerase, and 5 ul of the repair reaction, and ddH20 to 50 ul. The PCR cycle for SmaI/AatII was 95° C. for 5 minutes, followed by 25 cycles of 95° C. for 1 minute, 65° C. for 1 minute and 74° C. for 1 minute, with a final extension period of 10 minutes at 74° C. The cycle was the same for the HindIII/EcoRI reaction, but the annealing temperature used was 63° C., instead of 65° C.
[0132] 5 ul of the PCR reaction was run on a 1% agarose Et-Br gel, and visualised under UV light. The PCR products were compared with the product given when PCR was carried out on uncut plasmid, with repaired product showing a PCR band ˜400-600 bp smaller than that given by the PCR on the uncut plasmid. 5 ul of reactions showing successful repair was transformed into electro-competent XL1 blue cells, and resulting colonies were grown in 2×TY, plasmid clones purified and the repaired junctions sequenced.
Suicide Deletion Assays
[0133] The construction of the suicide deletion allele ade2::SD2 shown in FIG. 13A was as described in Karathanasis et al Genetics 161, 1015 (2002)). The gene conversion donor was constructed by PCR-mediated gene replacement of the CAN1 gene with a fragment of ADE2 that contains a 7-base insertion just downstream of the start codon, the same location as the I-SceI and HO sites in the suicide deletion cassettes. There was ˜650 by of ADE2 homology on each side of the cut site position. The HO suicide deletion allele shown in FIG. 13B was constructed by the same method used to create the ade2::SD2-allele (E. Karathanasis, T. E. Wilson, Genetics 161, 1015 (2002)), except amplifying the GAL1-HO cassette from pGAL-HO (T. E. Wilson, M. R. Lieber, J. Biol. Chem. 274, 23599 (1999)) and incorporating HO cut sites. The exact sequence of all alleles is available upon request. Strains were isogenic derivatives of S288C (C. B. Brachmann et al Yeast 14, 115 (1998)). yku70, dn14 and rad50 mutants were made by PCR-mediated gene replacement and multiple mutants thereof were made by mating and sporulation. The data shown in FIGS. 8 to 10 were generated by growth in glucose liquid medium followed by plating to galactose plates. Data are colony counts from galactose (either Ade.sup.+ or Ade.sup.-) divided by colony counts from parallel glucose plates. This method reveals the absolute frequency of simple religation NHEJ (Wilson, T. E. Genetics 162, 677 (2002)). The data in FIGS. 11 and 12 were generated by allowing cultures to grow out in non-selective galactose liquid medium prior to plating to glucose plates. Data in graphs are the ratio of Ade.sup.+ to total colonies. This method measures the frequency of imprecise NHEJ. All data points represent the average±standard deviation of at least 3 independent measurements.
Expression of Mt NHEJ Proteins in Yeast
[0134] Plasmids pNLS15 and pNLS16 are CEN plasmids (LEU2- and URA3-selectable, respectively) that direct the expression of cloned cDNAs in yeast as amino-terminal Myc epitope-NLS fusion proteins from the strong constitutive ADH1 promoter. These were made by PCR amplification of the ADH1 promoter and YKU70 terminator regions, subsequent PCR fusion via primer overhangs to generate the Myc-NLS linker region, and finally ligation into pRS415 and pRS416 (C. B. Brachmann et al Yeast 14, 115 (1998)). Mt Rv0937c and Rv0938 coding sequences were inserted into pNLS15 and pNLS16, respectively, by the gap repair technique. Briefly, the vectors were digested with Sma I and co-transformed into yeast with PCR fragments of the bacterial genes that contained 45 by 5' extensions flanking the Sma I site. Following mating and sporulation to facilitate suicide deletion screening, the plasmids from a functional Ku-ligase pair were recovered from yeast and sequenced to rule out unexpected mutations. These were re-transformed into fresh yeast in parallel with vectors as needed.
Fluorescent PCR of Yeast HO Joints
[0135] ˜108 cells from a 2-day yeast culture in glucose synthetic defined medium lacking uracil and leucine were inoculated into fresh 25 ml of the same medium with galactose as the carbon source. This culture was shaken at 30 C for 2 days, and then diluted back 30-fold into 25 ml fresh medium lacking adenine. Following an additional 2 days shaking, ˜6×107 cells were harvested and genomic DNA prepared. DNA (0.2 ug, 1.3×106 genome equivalents) was then used in a 20 μl PCR reaction with primers OW1708 (5'-HEX-CAAGTATGGATCTCGAGGTT) (SEQ ID NO: 22) and OW1709 (5'-CTGTTCTAGAGGTACCTAGT (SEQ ID NO: 23); 25 cycles of 94 C for 15 seconds and 55 C for 15 seconds). 2 μl was then run on an 8% sequencing gel.
Yeast Joint Analysis
[0136] All colonies analyzed for the nature of their repair event were independently derived. Colonies were purified by streaking and then colony PCR was performed using primers OW603 (5'-CCTTAAGTTGAACGGAGTCC) (SEQ ID NO: 24) and OW620 (5'-CTTGACTAGCGCACTACCAG) (SEQ ID NO: 25), which amplify a 1273 by fragment surrounding the HO or I-SceI cut sites in successful deletion events (the starting allele is too large to amplify). Recreated I-SceI sites were detected by cleavage in vitro with recombinant I-SceI (New England Biolabs) into the expected 574 and 699 by products. All other individual joint fragments were sequenced with primer OW563 (5'-GGCAGGAGAATTTTCAGCATC) (SEQ ID NO: 26) and their microhomology mediated joining mechanism inferred by comparison with an intact I-SceI or HO cut site.
Results
[0137] Mt-Ku Binding to DNA
[0138] Recombinant histidine-tagged versions of Mycobacterium tuberculosis Ku-like protein [open reading frame (ORF) Rv0937c] and the genetically linked putative ATP-dependent ligase (ORF Rv0938) were found to be readily over expressed in soluble form in E. coli. These proteins (designated Mt-Ku and Mt-Lig) were purified by nickel-agarose affinity chromatography.
[0139] Analysis of recombinant Mt-Ku by gel-filtration chromatography indicated that Mt-Ku exists as a homodimer in solution. This species was very stable, even at high salt concentrations, which provides indication of a strong homodimeric interaction. Electrophoretic mobility-shift assays (EMSAs), with a 33-base-pair (bp) dsDNA oligonucleotide with either 5' or 3' overhangs, demonstrated that Mt-Ku, like eukaryotic Ku, forms a specific complex with either type of DNA end. Excess non-labelled linear dsDNA, but not closed circular plasmid DNA or single-stranded DNA, competed for binding, which demonstrates that Mt-Ku binds preferentially to dsDNA ends.
[0140] Titration of Mt-Ku against fixed concentration of labelled 33-nucleotide oligomer resulted in a single retarded band, presumably representing a 1:1 Ku-DNA complex. When the length of the DNA was doubled (66-nucleotide oligomer), two progressively retarded bands were observed. Multiple Ku-DNA complexes were formed on all dsDNA linear substrates of >60-mer tested, and the number of retarded species was directly proportional to the length of the DNA, indicating that, after binding to the end, Mt-Ku can freely move along the DNA.
Mt-Lig Substrate
[0141] To test whether Mt-Lig uses ATP or NAD+, Mt-Lig was incubated with either [α-32P] ATP or NAD+ and magnesium. In the presence of ATP, but not NAD.sup.+, a radiolabelled covalent ligase-adenylate adduct was formed that co-migrated with the Mt-Lig polypeptide during SDS-polyacrylamide gel electrophoresis (SDS-PAGE). This demonstrates that Mt-Lig is active in covalent nucleotidyl transfer with a specific preference for ATP as the AMP donor.
[0142] Substitution of the motif I residue Lys481 by alanine (K481A) abolished ligase-AMP formation.
Ligase Activity of Mt-Lig
[0143] To examine whether Mt-Lig is a dsDNA ligase, dsDNA substrates of various sizes (53 to 2560 bp) were used in ligation reactions and the efficiency of ligation compared to that mediated by T4 DNA ligase. Mt-Lig catalyzed the joining of the various dsDNA fragments of different lengths to equivalent extents. M. tuberculosis Mt-Lig is therefore a functional DNA ligase capable of catalyzing DSB rejoining in an ATP-dependent manner.
[0144] Notably, the DNA ligation activity of Mt-Lig was stimulated >30-fold by the addition of Mt-Ku. Stimulation was abolished by heat denaturation of Mt-Ku. Mt-Lig was not stimulated by the human Ku heterodimer and, conversely, human ligase IV/XRCC4 and T4 ligase were not stimulated by Mt-Ku. Indeed, amounts of Mt-Ku that stimulated Mt-Lig inhibited both ligase IV and T4 ligase activity. Consistent with these observations, Mt-Ku stimulated the activity of Mt-Lig by 20-fold but not T4 ligase in an in vitro plasmid repair assay. Stimulation of ligation by Mt-Ku is therefore highly specific for Mt-Lig and provides indication that these proteins physically interact.
[0145] Potential interactions between Mt-Ku and Mt-Lig were investigated by EMSAs with a radiolabelled dsDNA probe (33 bp). Including Mt-Lig and Ku together led to the generation of a DNA/protein complex with a mobility distinct from that of the complexes formed by either protein alone. However, the addition of increasing amounts of Mt-Ku did not abolish the appearance of the novel DNA-protein complex, which demonstrates that Mt-Ku does not inhibit the binding of Mt-Lig to DNA. Formation of the new complex did not occur when Mt-Lig had been heat denatured, which indicates that the complex reflects the binding of Mt-Lig and is not mediated by a buffer component. Biacore studies with a biotinylated dsDNA (33-mer) bound to a streptavidin coated chip and isothermal titration calorimetry studies also confirmed that Mt-Ku specifically recruits Mt-Lig to DNA.
[0146] To determine whether Mt-Lig has RNA primase activity, recombinant Mt-Lig was incubated with a poly dT homopolymer and [α-32P] ATP. Mt-Lig was observed to synthesize oligoribonucleotides ranging in length from 1-50 nucleotides. In a similar assay with a single strand DNA template, Mt-Lig also synthesized RNA primers.
[0147] Mt-lig was assayed for DNA-dependent DNA primase activity using complementary single stranded oligonucleotides. Annealing of the complementary single-strands resulted in a 5-nt overhang in the bottom strand. Mt-Lig filled the overhangs with either dNTPs or rNTPs confirming the presence of both DNA-dependent DNA and RNA polymerase activities. Replacement of two invariant Asp residues in motif I of Mt-Lig with alanine residues abolished the polymerase activity of Mt-Lig.
[0148] Polymerisation assays were performed with DNA duplex oligonucleotides that generate a non-ligatable one nucleotide (nt) gap and a 5-base 3' overhang upon alignment (FIG. 4A). Mt-Lig efficiently filled in the gap with no detectable strand displacement synthesis (FIG. 5A, left panel). Addition of a phosphate group to the 5' terminus of the 1-nt gap, resulted in gap-filling and ligation (FIG. 5A, right panel), indicating the concerted action of Mt-Lig polymerase and ligase activities on NHEJ intermediates.
[0149] Mt-Lig progressively digested the 3' single-strands (ss) but not the 5' ss tails of partial duplexes until reaching the double-strand (ds) region (FIG. 5B). Thus, Mt-Lig possesses 3' to 5' ss DNA exonuclease activity. Using DNA substrates that generate a 3'-flap adjacent to a nick, Mt-Lig removed the flap by exonucleolytic digestion, generating a base-paired linear duplex (FIG. 5C). At higher concentrations the nuclease progressed through the microhomology region and into the duplex (FIG. 5C). Similar results were obtained when there was a gap adjacent to the mismatched flap. Nuclease activity was dependent on the presence of a divalent cation such as magnesium or manganese. Replacement of a conserved histidine residue (H373) with alanine abolished this exonuclease activity, confirming that the nuclease activity is also an intrinsic property of Mt-Lig.
[0150] The Mt-Lig complex was examined to see if it could repair a double strand break (DSB) junction containing non-compatible ends requiring full end processing prior to ligation.
[0151] In the presence of NTPs, Mt-Lig joined aligned DNA duplexes possessing a 1-nt 3' flap adjacent to 3-nt gap (FIG. 6A). A similar, albeit less efficient reaction, was observed in the presence of dNTPs. Neither the nuclease or polymerase mutant proteins were able to repair this junction, confirming that both activities are required to process the DSB prior to ligation. A synthetic DNA DSB junction was designed that contained a micro-homology (4 bp), a ssDNA gap (5 bp) and a 3' ssDNA flap structure (3 bp).
[0152] Sequencing of ligated junctions generated by Mt-Lig in assays with this substrate with a 3-nt flap adjacent to a 5-nt gap revealed that microhomology sequence was retained and the mismatched flap was replaced by nucleotides complementary to the template strand.
[0153] Mt-Lig was observed to be capable of removing the 3' flap overhang. However, the 3' processing activity also excised the micro-homology sequence back to the ds DNA junction. Similar processing activity was also observed on gapped, micro-homology substrates with no 3' flap. These findings confirm that Mt-Lig possesses a structure specific 3' exonuclease that removes 3' overhangs of DNA ends or DSBs.
[0154] Mt-Lig was assayed for both DNA and RNA "filling-in" activity on the micro-homology DSB substrate. Mt-Lig synthesized DNA or RNA, depending on the nucleotide added, and effectively filled in the 5 by gap.
[0155] The effect of Mt-Lig on DNA molecules with incompatible ends was assessed. In the presence of nucleotides (NTPs or dNTPs), ATP and magnesium, the three catalytic activities of Mt-Lig were observed to act in a concerted manner to selectively and precisely process DNA molecules with incompatible ends and join the resulting reconstituted compatible ends.
[0156] In the first step of Mt-Lig mediated ligation, the 3' nuclease activity cleaves away 7 bp (3 bp flap plus 4 bp micro-homology) leaving a dsDNA end. The nucleolysis step is followed by a polymerisation step to fill in the resulting gap, visible as a ladder of incompletely filled-in products. Finally, the fully extended strand is ligated to the 5' phosphate of the other DSB yielding one of the most abundant species, the fully ligated DSB. Sequencing of the repaired DSB junctions confirmed that the flap was removed and replaced with the sequence of the complementary template strand.
[0157] Mt-Ku specifically stimulated joining of fully complementary ss-ends by Mt-Lig as described above. The impact of Mt-Ku on the other activities of Mt-Lig was examined. Mt-Ku had no significant effect on the removal of mismatched flaps, but did inhibit further digestion into the microhomology region (FIG. 6B), providing indication that Mt-Ku remains physically associated with this region during repair.
[0158] The role of Mt-Ku was examined using an in vitro PCR-based plasmid repair assay (D. A. Ramsden et al Nature 388 488 (1997)) In this assay, plasmid DNA was cut with different pairs of restriction enzymes, incubated with Mt-Lig in the presence or absence of Mt-Ku, and finally the repaired DSB junction was amplified by PCR and sequenced. Mt-Ku was observed to dramatically stimulate joining of long linear DNA molecules with different incompatible ends by Mt-Lig (FIG. 6C). Processing and joining occurred in the presence of either dNTPs or NTPs (FIG. 6C). In contrast, no rejoining was observed by T4 ligase in the presence or absence of Mt-Ku (FIG. 6C). Joining of partially complementary 5' (HindIII-NheI) and 3' (PstI-KpnI) overhangs appeared to require microhomology-mediated alignments that need gap filling and, in some instances, 3' flap removal on one strand (Table 1). Joining of blunt end-3' single-strand overhang (SumI-AatII) appeared to require the addition of one nucleotide by the terminal transferase activity, followed by microhomology pairing with the 3' overhang, flap resection, gap filling, and ligation (Table 1). In all cases, gap-filling accurately copied the template strand.
[0159] These findings demonstrated that Mt Ku and ligase can perform NHEJ in vitro. To establish if the complex could mediate rejoining of chromosomal breaks in vivo, a variant of the yeast-based "suicide deletion" assay was employed (E. Karathanasis et al Genetics 161, 1015 (2002); Wilson, T. E., Genetics 162, 677 (2002)). This allowed the simultaneous determination of NHEJ and recombination frequencies.
[0160] ˜75% of wild-type yeast cells repaired the I-SceI DSB by recombination and ˜2% by NHEJ, with the remainder dying (FIG. 8). NHEJ occurred predominantly by simple religation (Ade.sup.+ colonies) and was -100-fold decreased by yku70 (Ku) deletion. Introducing plasmids expressing Mt-Ku and Mt-Lig restored NHEJ to ˜50% of the wild-type yeast level (FIG. 8). The pattern seen with combinations of Mt-Ku, Mt-Lig and yku70 and dn14 (ligase) mutations demonstrated that Mt NHEJ was truly reconstituted by a concerted species-specific interaction of the Ku and ligase proteins independent of yeast NHEJ (FIG. 9).
[0161] In the yeast S. cerevisiae, NHEJ is also dependent upon the Mre11/Rad50/Xrs2 complex (MRX). MRX may act as an end-bridging factor and/or functionally interact with yeast Ku and Dn14/Lift. Expression of the Mt NHEJ proteins in yeast rad50 mutants substantially recovered NHEJ (FIG. 10), although to a lesser extent than seen with yku70 or dn14 mutants. Thus, Mt NHEJ reconstitution in yeast required neither MRX nor its bacterial orthologue SbcCD, demonstrating that MRX-family function is not obligatorily required for tethering of chromosome ends during NHEJ.
[0162] As with NHEJ mediated by yeast proteins (T. E. Wilson et al Nature 388, 495-498 (1997)), Mt NHEJ reconstituted in yeast occasionally resulted in imperfect repair, evident as Ade.sup.- colonies in the absence of the gene conversion donor. Sequencing 15 of these colonies revealed a variety of junctions that occurred predominantly by mispairing of the A/T-rich I-SceI 3' overhangs.
[0163] To create a suicide deletion system that selects specifically for NHEJ events involving such end processing, HO was substituted for I-SceI so that +2 (or -1, -4, etc.) frame-shifted joints yield Ade.sup.+ colonies. ˜0.75% of all NHEJ events in wild-type yeast were Ade.sup.+ (FIG. 11), and >50% of these were HO(+2) joints (FIG. 11). With Mt NHEJ reconstituted, the overall frequency of NHEJ remained high, but the percentage of Ade.sup.+ events was substantially decreased (FIG. 11). Although some HO(+2) processed joints were formed, the HO(-1) joint now predominated (FIG. 12), providing a signature for Mt NHEJ. Strikingly, Mt NHEJ proteins shifted the HO joint pattern and Ade.sup.+ frequency to match that observed for Mt NHEJ even in wild-type yeast (FIG. 11). Mt-Ku and Mt-Lig proteins can therefore catalyze processed NHEJ in chromosomes, but, despite this ability, repair is highly accurate at compatible DSB ends.
[0164] The above findings demonstrate that Mt-Lig possesses the nuclease, ligase and polymerase activities which are required for non-homologous end joining (NHEJ). NHEJ repair assays further show that the activities of this polypeptide act in a concerted manner to selectively and precisely process DNA molecules with incompatible ends and join the resulting reconstituted compatible ends, allowing the NHEJ pathway to be reconstituted in vitro and in vivo using Mt-Lig.
TABLE-US-00001 No. of Non-homologus ends clones Predicted intermediates Repaired NHEJ junction ##STR00001## 10 ##STR00002## ##STR00003## 1 ##STR00004## ##STR00005## ##STR00006## 10 ##STR00007## ##STR00008## 2 ##STR00009## ##STR00010## ##STR00011## 6 ##STR00012## ##STR00013## 8 ##STR00014## ##STR00015##
TABLE-US-00002 TABLE 2 Bs ykoU: 21 EVKYDGYR 43 LTLDGEIV 34 CFLAFDLLERSG 57 EGIVA 15 WLKYKNFKQAY 82 IGFEFQMDWTE 304 (SEQ ID (SEQ ID (SEQ ID (SEQ ID (SEQ ID (SEQ ID NO: 27) NO: 28) NO: 29) NO: 30) NO: 31) NO: 32) Bh 2209: 20 EVKYDGFR 43 ITIDGELV 34 TLLAFDILELKG 57 EGVVA 15 WLKKKNFRQVT 81 HRFRLDVKPAQ 306 (SEQ ID (SEQ ID (SEQ ID (SEQ ID (SEQ ID (SEQ ID NO: 33) NO: 34) NO: 35) NO: 36) NO: 37) NO: 38) Mt Lig C: 26 EPKYDGFR 38 CVIDGEII 32 SFIAFDLLALGD 54 DGVIA 13 MFKIKHLRTAD 114 TAQFNRWRPDR 26 (SEQ ID (SEQ ID (SEQ ID (SEQ ID (SEQ ID (SEQ ID NO: 39) NO: 40) NO: 41) NO: 42) NO: 43) NO: 44) Bs yoqV: 22 ELKYDGIR 35 TVLDGEVI 26 VYCVFDVIYKDG 47 EGIVI 15 WLKVINYDYTE 81 (SEQ ID (SEQ ID (SEQ ID (SEQ ID (SEQ ID (SEQ ID NO: 45) NO: 46) NO: 47) NO: 48) NO: 49) NO: 50) Pa 2138: 235 ELKYDGYR 38 SWLDGELV 35 LYVLFDLPYHEG 49 EGVIG 14 WIKLKCQLRQE 111 AREVTGERPAG 313 (SEQ ID (SEQ ID (SEQ ID (SEQ ID (SEQ ID (SEQ ID NO: 50) NO: 51) NO: 52) NO: 53) NO: 54) NO: 55) Mt Rv0938 478 EGKYDGYR 38 VVLDGEAV 22 EFWAFDLLYLDG 46 EGVIA 15 WVKDKHWNTQE 98 -SSWRGLRPDK 8 (SEQ ID (SEQ ID (SEQ ID (SEQ ID (SEQ ID (SEQ ID NO: 56) NO: 57) NO: 58) NO: 59) NO: 60) NO: 61) Bact ATP s. KhDGhr ..hpGEhh .h.hFDh....s eghhh hhK.K...... ........... Consensus: (SEQ ID (SEQ ID (SEQ ID (SEQ ID (SEQ ID (SEQ ID NO: 62) NO: 63) NO: 64) NO: 65) NO: 66) NO: 67) EIKYDGvR FMLDGELM HIKLYAILPL-- 62 EGLIV 14 WWKMKPENEAD 96 PSFWM-FRGTE 7 T7 Lig: 31 (SEQ ID 48 (SEQ ID 49 (SEQ ID (SEQ ID (SEQ ID (SEQ ID NO: 66) NO: 67) NO: 68) NO: 69) NO: 70) NO: 71) Motif I III IIIa IV V VI
Sequence CWU
1
92110PRTArtificial sequenceConserved motif 1Arg Leu Val Phe Asp Leu Asp
Pro Gly Glu1 5 10210PRTArtificial
sequenceConserved motif 2Ser Gly Ser Lys Gly Leu His Leu Tyr Thr1
5 1036PRTArtificial sequenceConserved motif 3Lys
Val Phe Val Asp Trp1 5420DNAArtificial sequencePrimer
4atgcgagcca tttggacggg
20525DNAArtificial sequencePrimer 5ggatcctcac ggaggcgttg ggacg
25620DNAArtificial sequencePrimer
6atgggttcgg cgtcggagca
20726DNAArtificial sequencePrimer 7tcctcattcg cgcaccacct cactgg
26815DNAArtificial sequenceOligonucleotide
8ctgcagctga tgcgc
15920DNAArtificial sequenceOligonucleotide 9atccggcgca tcagctgcag
201025DNAArtificial
sequenceOligonucleotide 10agtcgatcct gcgcatcatc tgcag
251141DNAArtificial sequenceOligonucleotide
11acccggggat ccgtacagtc tatccggcgc atcagctgca g
411250DNAArtificial sequence50mer substrate 12gtaacaaagt ttggattgct
actgaccgct ctcgtgctcg tcgctgcgtt 501350DNAArtificial
sequenceOligonucleotide 13gtctgtctca ctattagaac cctttagagt catgcgtcgc
gaggcaacgc 501443DNAArtificial sequenceOligonucleotide
14gcctcgcgac gcatgactct aaagggttct aatagtgaga cag
431541DNAArtificial sequenceOligonucleotide 15gcgacgagca cgagagcggt
cagtagcaat ccaaactttg t 411650DNAArtificial
sequenceOligonucleotide 16gtaacaaagt ttggattgct actgaccgct ctcgtgctcg
tcgctgcgtt 501752DNAArtificial sequenceOligonucleotide
17ctgtctgtct cactattaga accctttaga gtcatgcgtc gcgaggcaac gc
521820DNAArtificial sequenceOligonucleotide 18gaaaccacgt accggcgtgt
201913DNAArtificial
sequenceOligonucleotide 19ctttggtcga tgg
132026DNAArtificial sequenceOligonucleotide
20ctgcagatca tgcgccggat tgcccc
262117DNAArtificial sequenceOligonucleotide 21gacgtctagt acgcggc
172220DNAArtificial
sequencePrimer 22caagtatgga tctcgaggtt
202320DNAArtificial sequencePrimer 23ctgttctaga ggtacctagt
202420DNAArtificial
sequencePrimer 24ccttaagttg aacggagtcc
202520DNAArtificial sequencePrimer 25cttgactagc gcactaccag
202621DNAArtificial
sequencePrimer 26ggcaggagaa ttttcagcat c
21278PRTBacillus subtilis 27Glu Val Lys Tyr Asp Gly Tyr Arg1
5288PRTBacillus subtilis 28Leu Thr Leu Asp Gly Glu Ile Val1
52912PRTBacillus subtilis 29Cys Phe Leu Ala Phe Asp Leu Leu
Glu Arg Ser Gly1 5 10305PRTBacillus
subtilis 30Glu Gly Ile Val Ala1 53111PRTBacillus subtilis
31Trp Leu Lys Tyr Lys Asn Phe Lys Gln Ala Tyr1 5
103211PRTBacillus subtilis 32Ile Gly Phe Glu Phe Gln Met Asp Trp
Thr Glu1 5 10338PRTBacillus halodurans
33Glu Val Lys Tyr Asp Gly Phe Arg1 5348PRTBacillus
halodurans 34Ile Thr Ile Asp Gly Glu Leu Val1
53512PRTBacillus halodurans 35Thr Leu Leu Ala Phe Asp Ile Leu Glu Leu Lys
Gly1 5 10365PRTBacillus halodurans 36Glu
Gly Val Val Ala1 53711PRTBacillus halodurans 37Trp Leu Lys
Lys Lys Asn Phe Arg Gln Val Thr1 5
103811PRTBacillus halodurans 38His Arg Phe Arg Leu Asp Val Lys Pro Ala
Gln1 5 10398PRTMycobacterium tuberculosis
39Glu Pro Lys Trp Asp Gly Phe Arg1 5408PRTMycobacterium
tuberculosis 40Cys Val Ile Asp Gly Glu Ile Ile1
54112PRTMycobacterium tuberculosis 41Ser Phe Ile Ala Phe Asp Leu Leu Ala
Leu Gly Asp1 5 10425PRTMycobacterium
tuberculosis 42Asp Gly Val Ile Ala1 54311PRTMycobacterium
tuberculosis 43Met Phe Lys Ile Lys His Leu Arg Thr Ala Asp1
5 104411PRTMycobacterium tuberculosis 44Thr Ala Gln Phe
Asn Arg Trp Arg Pro Asp Arg1 5
10458PRTBacillus subtilis 45Glu Leu Lys Phe Asp Gly Ile Arg1
5468PRTBacillus subtilis 46Thr Val Leu Asp Gly Glu Val Ile1
54712PRTBacillus subtilis 47Val Tyr Cys Val Phe Asp Val Ile Tyr Lys Asp
Gly1 5 10485PRTBacillus subtilis 48Glu
Gly Ile Val Ile1 54911PRTBacillus subtilis 49Trp Leu Lys
Val Ile Asn Tyr Asp Tyr Thr Glu1 5
10508PRTPseudomonas aeruginosa 50Glu Leu Lys Leu Asp Gly Tyr Arg1
5518PRTPseudomonas aeruginosa 51Ser Trp Leu Asp Gly Glu Leu Val1
55212PRTPseudomonas aeruginosa 52Leu Tyr Val Leu Phe Asp Leu
Pro Tyr His Glu Gly1 5
10535PRTPseudomonas aeruginosa 53Glu Gly Val Ile Gly1
55411PRTPseudomonas aeruginosa 54Trp Ile Lys Leu Lys Cys Gln Leu Arg Gln
Glu1 5 105511PRTPseudomonas aeruginosa
55Ala Arg Glu Val Thr Gly Glu Arg Pro Ala Gly1 5
10568PRTMycobacterium tuberculosis 56Glu Gly Lys Trp Asp Gly Tyr
Arg1 5578PRTMycobacterium tuberculosis 57Val Val Leu Asp
Gly Glu Ala Val1 55812PRTMycobacterium tuberculosis 58Glu
Phe Trp Ala Phe Asp Leu Leu Tyr Leu Asp Gly1 5
10595PRTMycobacterium tuberculosis 59Glu Gly Val Ile Ala1
56011PRTMycobacterium tuberculosis 60Trp Val Lys Asp Lys His Trp Asn
Thr Gln Glu1 5 106110PRTMycobacterium
tuberculosis 61Ser Ser Trp Arg Gly Leu Arg Pro Asp Lys1 5
10626PRTArtificial sequenceBact ATP Consensus 62Lys Xaa
Asp Gly Xaa Arg1 5636PRTArtificial sequenceBact ATP
Consensus 63Xaa Xaa Gly Glu Xaa Xaa1 5644PRTArtificial
sequenceBact ATP Consensus 64Xaa Phe Asp Xaa1655PRTArtificial
sequenceBact ATP Consensus 65Glu Gly Xaa Xaa Xaa1 5668PRTT7
66Glu Ile Lys Tyr Asp Gly Val Arg1 5678PRTT7 67Phe Met Leu
Asp Gly Glu Leu Met1 56810PRTT7 68His Ile Lys Leu Tyr Ala
Ile Leu Pro Leu1 5 10695PRTT7 69Glu Gly
Leu Ile Val1 57011PRTT7 70Trp Trp Lys Met Lys Pro Glu Asn
Glu Ala Asp1 5 107110PRTT7 71Pro Ser Phe
Val Met Phe Arg Gly Thr Glu1 5
107210PRTMycobacterium tuberculosis 72Arg Leu Val Phe Asp Leu Asp Pro Gly
Glu1 5 107310PRTMycobacterium
tuberculosis 73Ser Gly Ser Lys Gly Leu His Leu Tyr Thr1 5
10746PRTMycobacterium tuberculosis 74Lys Val Phe Val Asp
Trp1 57510PRTHomo sapiens 75Glu Leu Val Phe Asp Ile Asp Met
Thr Asp1 5 107610PRTHomo sapiens 76Ser
Gly Arg Arg Gly Val His Cys Trp Val1 5
10776PRTHomo sapiens 77Phe Pro Arg Leu Asp Ile1 5786PRTMus
musculus 78Phe Pro Arg Leu Asp Val1 57910PRTDrosophila
melanogaster 79Ser Gly Arg Arg Gly Ile His Cys Trp Val1 5
10806PRTDrosophila melanogaster 80Tyr Pro Arg Leu Asp
Ile1 58110PRTSaccharomyces cerevisiae 81Glu Leu Val Phe Asp
Ile Asp Met Asp Asp1 5
108210PRTSaccharomyces cerevisiae 82Ser Gly Arg Arg Gly Ala His Cys Trp
Val1 5 10836PRTSaccharomyces cerevisiae
83Tyr Pro Lys Leu Asp Val1 58410PRTSchizosaccharomyces
pombe 84Ser Gly Arg Arg Gly Ile His Ala Trp Ile1 5
10856PRTSchizosaccharomyces pombe 85Tyr Pro Arg Leu Asp Val1
58613DNAArtificial sequenceSynthetic oligomer 86ggtacgtggt ttc
138717DNAArtificial
sequenceSynthetic oligonucleotide 87cggcgcatga tctgcag
178820DNAArtificial sequenceSynthetic
oligonucleotide 88gaaaccacgt accggggtgt
208939DNAArtificial sequenceSynthetic oligomer 89ctgcagatca
tgcgccggat tgccccggta cgtggtttc
399039DNAArtificial sequenceSynthetic oligonucleotide 90gaaaccacgt
accggggcaa tccggcgcat gatctgcag
3991759PRTMycobacterium tuberculosis 91Met Gly Ser Ala Ser Glu Gln Arg
Val Thr Leu Thr Asn Ala Asp Lys1 5 10
15Val Leu Tyr Pro Ala Thr Gly Thr Thr Lys Ser Asp Ile Phe
Asp Tyr 20 25 30Tyr Ala Gly
Val Ala Glu Val Met Leu Gly His Ile Ala Gly Arg Pro 35
40 45Ala Thr Arg Lys Arg Trp Pro Asn Gly Val Asp
Gln Pro Ala Phe Phe 50 55 60Glu Lys
Gln Leu Ala Leu Ser Ala Pro Pro Trp Leu Ser Arg Ala Thr65
70 75 80Val Ala His Arg Ser Gly Thr
Thr Thr Tyr Pro Ile Ile Asp Ser Ala 85 90
95Thr Gly Leu Ala Trp Ile Ala Gln Gln Ala Ala Leu Glu
Val His Val 100 105 110Pro Gln
Trp Arg Phe Val Ala Glu Pro Gly Ser Gly Glu Leu Asn Pro 115
120 125Gly Pro Ala Thr Arg Leu Val Phe Asp Leu
Asp Pro Gly Glu Gly Val 130 135 140Met
Met Ala Gln Leu Ala Glu Val Ala Arg Ala Val Arg Asp Leu Leu145
150 155 160Ala Asp Ile Gly Leu Val
Thr Phe Pro Val Thr Ser Gly Ser Lys Gly 165
170 175Leu His Leu Tyr Thr Pro Leu Asp Glu Pro Val Ser
Ser Arg Gly Ala 180 185 190Thr
Val Leu Ala Lys Arg Val Ala Gln Arg Leu Glu Gln Ala Met Pro 195
200 205Ala Leu Val Thr Ser Thr Met Thr Lys
Ser Leu Arg Ala Gly Lys Val 210 215
220Phe Val Asp Trp Ser Gln Asn Ser Gly Ser Lys Thr Thr Ile Ala Pro225
230 235 240Tyr Ser Leu Arg
Gly Arg Thr His Pro Thr Val Ala Ala Pro Arg Thr 245
250 255Trp Ala Glu Leu Asp Asp Pro Ala Leu Arg
Gln Leu Ser Tyr Asp Glu 260 265
270Val Leu Thr Arg Ile Ala Arg Asp Gly Asp Leu Leu Glu Arg Leu Asp
275 280 285Ala Asp Ala Pro Val Ala Asp
Arg Leu Thr Arg Tyr Arg Arg Met Arg 290 295
300Asp Ala Ser Lys Thr Pro Glu Pro Ile Pro Thr Ala Lys Pro Val
Thr305 310 315 320Gly Asp
Gly Asn Thr Phe Val Ile Gln Glu His His Ala Arg Arg Pro
325 330 335His Tyr Asp Phe Arg Leu Glu
Cys Asp Gly Val Leu Val Ser Trp Ala 340 345
350Val Pro Lys Asn Leu Pro Asp Asn Thr Ser Val Asn His Leu
Ala Ile 355 360 365His Thr Glu Asp
His Pro Leu Glu Tyr Ala Thr Phe Glu Gly Ala Ile 370
375 380Pro Ser Gly Glu Tyr Gly Ala Gly Lys Val Ile Ile
Trp Asp Ser Gly385 390 395
400Thr Tyr Asp Thr Glu Lys Phe His Asp Asp Pro His Thr Gly Glu Val
405 410 415Ile Val Asn Leu His
Gly Gly Arg Ile Ser Gly Arg Tyr Ala Leu Ile 420
425 430Arg Thr Asn Gly Asp Arg Trp Leu Ala His Arg Leu
Lys Asn Gln Lys 435 440 445Asp Gln
Lys Val Phe Glu Phe Asp Asn Leu Ala Pro Met Leu Ala Thr 450
455 460His Gly Thr Val Ala Gly Leu Lys Ala Ser Gln
Trp Ala Phe Glu Gly465 470 475
480Lys Trp Asp Gly Tyr Arg Leu Leu Val Glu Ala Asp His Gly Ala Val
485 490 495Arg Leu Arg Ser
Arg Ser Gly Arg Asp Val Thr Ala Glu Tyr Pro Gln 500
505 510Leu Arg Ala Leu Ala Glu Asp Leu Ala Asp His
His Val Val Leu Asp 515 520 525Gly
Glu Ala Val Val Leu Asp Ser Ser Gly Val Pro Ser Phe Ser Gln 530
535 540Met Gln Asn Arg Gly Arg Asp Thr Arg Val
Glu Phe Trp Ala Phe Asp545 550 555
560Leu Leu Tyr Leu Asp Gly Arg Ala Leu Leu Gly Thr Arg Tyr Gln
Asp 565 570 575Arg Arg Lys
Leu Leu Glu Thr Leu Ala Asn Ala Thr Ser Leu Thr Val 580
585 590Pro Glu Leu Leu Pro Gly Asp Gly Ala Gln
Ala Phe Ala Cys Ser Arg 595 600
605Lys His Gly Trp Glu Gly Val Ile Ala Lys Arg Arg Asp Ser Arg Tyr 610
615 620Gln Pro Gly Arg Arg Cys Ala Ser
Trp Val Lys Asp Lys His Trp Asn625 630
635 640Thr Gln Glu Val Val Ile Gly Gly Trp Arg Ala Gly
Glu Gly Gly Arg 645 650
655Ser Ser Gly Val Gly Ser Leu Leu Met Gly Ile Pro Gly Pro Gly Gly
660 665 670Leu Gln Phe Ala Gly Arg
Val Gly Thr Gly Leu Ser Glu Arg Glu Leu 675 680
685Ala Asn Leu Lys Glu Met Leu Ala Pro Leu His Thr Asp Glu
Ser Pro 690 695 700Phe Asp Val Pro Leu
Pro Ala Arg Asp Ala Lys Gly Ile Thr Tyr Val705 710
715 720Lys Pro Ala Leu Val Ala Glu Val Arg Tyr
Ser Glu Trp Thr Pro Glu 725 730
735Gly Arg Leu Arg Gln Ser Ser Trp Arg Gly Leu Arg Pro Asp Lys Lys
740 745 750Pro Ser Glu Val Val
Arg Glu 75592273PRTMycobacterium tuberculosis 92Met Arg Ala Ile
Trp Thr Gly Ser Ile Ala Phe Gly Leu Val Asn Val1 5
10 15Pro Val Lys Val Tyr Ser Ala Thr Ala Asp
His Asp Ile Arg Phe His 20 25
30Gln Val His Ala Lys Asp Asn Gly Arg Ile Arg Tyr Lys Arg Val Cys
35 40 45Glu Ala Cys Gly Glu Val Val Asp
Tyr Arg Asp Leu Ala Arg Ala Tyr 50 55
60Glu Ser Gly Asp Gly Gln Met Val Ala Ile Thr Asp Asp Asp Ile Ala65
70 75 80Ser Leu Pro Glu Glu
Arg Ser Arg Glu Ile Glu Val Leu Glu Phe Val 85
90 95Pro Ala Ala Asp Val Asp Pro Met Met Phe Asp
Arg Ser Tyr Phe Leu 100 105
110Glu Pro Asp Ser Lys Ser Ser Lys Ser Tyr Val Leu Leu Ala Lys Thr
115 120 125Leu Ala Glu Thr Asp Arg Met
Ala Ile Val His Phe Thr Leu Arg Asn 130 135
140Lys Thr Arg Leu Ala Ala Leu Arg Val Lys Asp Phe Gly Lys Arg
Glu145 150 155 160Val Met
Met Val His Thr Leu Leu Trp Pro Asp Glu Ile Arg Asp Pro
165 170 175Asp Phe Pro Val Leu Asp Gln
Lys Val Glu Ile Lys Pro Ala Glu Leu 180 185
190Lys Met Ala Gly Gln Val Val Asp Ser Met Ala Asp Asp Phe
Asn Pro 195 200 205Asp Arg Tyr His
Asp Thr Tyr Gln Glu Gln Leu Gln Glu Leu Ile Asp 210
215 220Thr Lys Leu Glu Gly Gly Gln Ala Phe Thr Ala Glu
Asp Gln Pro Arg225 230 235
240Leu Leu Asp Glu Pro Glu Asp Val Ser Asp Leu Leu Ala Lys Leu Glu
245 250 255Ala Ser Val Lys Ala
Arg Ser Lys Ala Asn Ser Asn Val Pro Thr Pro 260
265 270Pro
User Contributions:
Comment about this patent or add new information about this topic:
 |  |
 |  |
 |  |
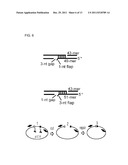 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
| New patent applications in this class: | |
| Date | Title |
|---|---|
| 2016-06-30 | Enhanced ligation reactions |
| 2016-06-30 | Thermostable blunt-end ligase and methods of use |
| 2016-03-03 | Synthon formation |
| 2016-01-28 | Nucleic acid binding proteins and uses thereof |
| 2016-01-28 | Methods for amplification of nucleic acids utilizing a circularized template prepared from a target nucleic acid |
| New patent applications from these inventors: | |
| Date | Title |
|---|---|
| 2012-05-31 | Dna damage repair inhibitors for the treatment of cancer |
| Top Inventors for class "Chemistry: molecular biology and microbiology" | |
| Rank | Inventor's name |
|---|---|
| 1 | Marshall Medoff |
| 2 | Anthony P. Burgard |
| 3 | Mark J. Burk |
| 4 | Robin E. Osterhout |
| 5 | Rangarajan Sampath |