Patent application title: METHODS AND MATERIALS RELATING TO GENE EXPRESSION
Inventors:
Keith Frederick Chater (Norwich, GB)
Celia Joyce Bruton (Norwich, GB)
Sean Joseph O'Rourke (Cork, IE)
Andreas Wilhelm Wietzorrek (Bodelshausen, DE)
IPC8 Class: AC12P2104FI
USPC Class:
435 691
Class name: Chemistry: molecular biology and microbiology micro-organism, tissue cell culture or enzyme using process to synthesize a desired chemical compound or composition recombinant dna technique included in method of making a protein or polypeptide
Publication date: 2008-10-30
Patent application number: 20080268501
Inventors list |
Agents list |
Assignees list |
List by place |
Classification tree browser |
Top 100 Inventors |
Top 100 Agents |
Top 100 Assignees |
Usenet FAQ Index |
Documents |
Other FAQs |
Patent application title: METHODS AND MATERIALS RELATING TO GENE EXPRESSION
Inventors:
Keith Frederick Chater
Celia Joyce Bruton
Sean Joseph O'Rourke
Andreas Wilhelm Wietzorrek
Agents:
DANN, DORFMAN, HERRELL & SKILLMAN
Assignees:
Origin: PHILADELPHIA, PA US
IPC8 Class: AC12P2104FI
USPC Class:
435 691
Abstract:
An expression cassette for expressing a nucleic acid of interest derived
from the regulatory region of the methylenomycin gene cluster of the SCP1
plasmid of Streptomyces coelicolor A3(2), and related materials and
methods.Claims:
1.-50. (canceled)
51. An expression product produced by a method comprising:(1) providing an expression system comprising an expression cassette for the expression of a nucleic acid of interest, said expression cassette including:a. at least one regulatory portion including:i. a first regulatory element which includes at least one of the nucleic acid sequences mmyR encoding an MmyR polypeptide, and mmfR encoding an MmfR polypeptide;ii. a second regulatory element which includes the nucleic acid sequence mmfL encoding an MmfL polypeptide; andiii. a promoter (the "repressible promoter"), the function of which is repressed by the expression product of the first regulatory element, that repression being alleviated or removed by a product, the production of which is conferred by the MmfL polypeptide, andb. a heterologous nucleic acid of interest, in operative association with said promoter; and(2) maintaining said expression system under conditions suitable for expression of the nucleic acid of interest.
52. (canceled)
53. A nucleic acid consisting essentially of at least one of the nucleic acid sequences mmyR, mmfP, mmfH, mmfL, mmfR, mmyT, mmyO and mmyG, optionally with at least one respective upstream region.
54. A nucleic acid according to claim 53, wherein said at least one nucleic acid sequence is selected from the following:a. the mmyR nucleic acid sequence encoding an MmyR polypeptide having the amino sequence of SEQ ID NO: 12 (optionally excluding the first 6 listed amino acids), or a variant thereof;b. the mmfR nucleic acid sequence encoding an MmfR polypeptide having the amino acid sequence of SEQ ID NO: 11, or a variant thereof;c. the mmfL nucleic acid sequence encoding an MmfL polypeptide having the amino acid sequence of SEQ ID NO: 17, or a variant thereof;d. the mmfP nucleic acid sequence encoding an MmfP polypeptide having the amino acid sequence of SEQ ID NO: 20, or a variant thereof;e. the mmfH nucleic acid sequence encoding an MmfH polypeptide having the amino acid sequence of SEQ ID NO: 21, or a variant thereof;f. the mmyT nucleic acid sequence encoding an MmyT polypeptide having the amino acid sequence of SEQ ID NO: 22 or a variant thereof;g. the mmyO nucleic acid sequence encoding an MmyO polypeptide having the amino acid sequence of SEQ ID NO: 23, or a variant thereof;h. the mmyG nucleic acid sequence encoding an MmyG polypeptide having the amino acid sequence of SEQ ID NO: 24, or a variant thereof; andi. the mmyB nucleic acid sequence comprising an MmyB polypeptide having the amino acid sequence set out in EMBL AJ276673, or a variant thereof.
55. A nucleic acid according to claim 53, wherein the upstream region comprises a promoter.
56. A nucleic acid according to claim 55, wherein the promoter is selected from the group consisting of:a. the mmyTOG promoter comprising at least some of residues 5452 to 5675 of SEQ ID NO: 18, or a variant thereof;b. the mmfLHP promoter comprising at least some of residues 4716 to 4909 of SEQ ID NO: 19, or a variant thereof;c. the mmy . . . XCAPK promoter comprising at least some of the complement of residues 18892 to 19123 or 15404 to 15977 of EMBL AJ276673, or a variant thereof;d. the mmyYF promoter comprising at least some of residues 18892 to 19123 of EMBL AJ276673;e. the mmyBQE promoter comprising at least some of the complement of residues 18892 to 19123 of EMBL AJ276673;f. the mmyR promoter comprising at least some of residues 7965 to 8132 of SEQ ID NO: 19 (optionally excluding residues 8113 to 8132), or a variant thereof;g. the mmfR promoter comprises at least some of residues 4613 to 4806 of SEQ ID NO: 18, or a variant thereof; andh. the mmfL promoter comprising at least some of residues 4716 to 4909 of SEQ ID NO: 19, or a variant thereof.
57. (canceled)
58. A polypeptide encodable by one of the nucleic acid sequences mmyR, mmfP, mmfH, mmfL, mmfR, mmyT, mmyO and mmyG.
59. A polypeptide according to claim 58 having an amino acid sequence as shown in one of FIGS. 8a to 8h, or a variant thereof.
60. A vector including a nucleic acid according to claim 53.
61. An expression system containing one or more nucleic acid according to claim 53.
62. A method of producing a polypeptide encodable by one of the nucleic acid sequences mmyR, mmfP, mmfH, mmfL, mmfR, mmyT, mmyO and mmyG, the method comprising maintaining an expression system as defined in claim 61 under conditions suitable for producing the polypeptide.
63. A method comprising providing a polypeptide produced according to the method of claim 62 and subjecting the polypeptide to one or more purification steps.
64. A nucleic acid according to claim 55, wherein the promoter is at least one of an mmfL promoter and an mmfR promoter comprising a palindromic sequence having the half-site 5'-GG(T/C)CGGT(A/T) (T/C)G(T/G)-3' (SEQ ID NO: 1), or a variant thereof having sequence identity at seven or more corresponding positions within the half-site.
65. An expression cassette according to claim 64, wherein the palindromic sequence has the half-site 5'-GGAAGGTATTA-3' (SEQ ID NO: 2), or a variant thereof.
Description:
[0001]The present invention relates to material derived from the SCP1
plasmid of Streptomyces coelicolor A3 (2) and methods and uses relating
thereto, in particular to material derived from the gene cluster for
methylenomycin A biosynthesis.
[0002]Underlying the invention is work carried out by the inventors in sequencing and deducing the function of various genes in the methylenomycin A biosynthetic gene cluster.
[0003]The natural role of the DNA to which the present invention relates is the production of the antibiotic methylenomycin A and its congeners. The whole cluster of methylenomycin production, resistance and regulatory genes (the mmy gene cluster) is known only from studies of Streptomyces coelicolor A3(2) and Streptomyces violaceoruber No. 2416 SANK 95570 (Chater and Bruton, 1985). In these two bacteria the genes concerned with methylenomycin production are present on different plasmids, SCP1 and pSV1 (Aguilar and Hopwood, 1982). No other example is known of plasmid-specified antibiotic production in Streptomyces. Where studied, all naturally occurring Streptomyces plasmids, including SCP1, can be transferred to new Streptomyces hosts by conjugation.
[0004]The DNA sequence of a 9.5 kb stretch of this gene cluster has now been discovered, and the inventors have identified several genes and their transcriptional organisation. They found that the transcriptional organisation of this region is significantly different from that suggested previously, for example in Chater and Bruton (1985). The function of certain of the genes has newly been deduced from the discovery that they display high levels of homology with other genes which are involved in the regulation of other, chromosomally located, antibiotic biosynthetic gene clusters in diverse streptomycetes.
[0005]This discovery is particularly surprising in view of the fact that methylenomycin is the only Streptomyces antibiotic whose biosynthesis is known to be conferred by a plasmid, rather than native Streptomyces genomic DNA. It implies that these genes should be adapted to function in an appropriately controlled manner in any Streptomyces host to which these plasmids may be transmitted.
[0006]Further, the inventors have discovered that the insertion of a gene of interest into a particular transcriptional unit within this 9.5 kb stretch allows that gene to be regulated so as to be expressed only at high cell density. This transcriptional unit contains three methylenomycin biosynthetic genes. Similar results were obtained for another transcriptional unit of the mmy gene cluster, indicating that other biosynthetic genes are similarly regulated, as well as for a transcriptional unit that is itself part of the regulatory system.
[0007]On the basis of these discoveries, the inventors now provide a model of how methylenomycin expression is regulated in Streptomyces.
[0008]This model allows predictions to be made about the effect of disrupting certain portions of the gene cluster. Observations consistent with these predictions have been made.
[0009]The inventors have sequenced and identified a block of eight genes, designated mmyR, mmfP, mmfH, mmfL, mmfR, mmyT, mmyO and mmyG. The arrangement of these genes in the sequenced stretch of DNA is shown in FIG. 1d. FIGS. 1a-c show the location of this stretch in the methylenomycin gene cluster and on the SCP1 plasmid.
[0010]The authors have further sequenced and identified five more genes, designated mmyK, mmyP, mmyA, mmyC and mmyX, which are part of an incompletely defined transcription unit, mmy . . . XCAPK, in a nearby block. The organisation of these genes, and further nearby genes, is shown in FIG. 1e. The sequence of this whole region, along with deduced amino acid sequences of the gene products, is now available in the GenBank/EMBL database, under accession number AJ276673.
[0011]At the heart of the system are the products of the two genes mmyR and mmfR. The inventors have discovered that these two genes encode proteins with very significant similarity to several other proteins from various Streptomyces spp. (FIG. 2). A known model member of this protein sub-family is ArpA, a protein of Streptomyces griseus (see e.g. Onaka and Horinouchi, 1997; Onaka et al., 1997; and Sugiyama et al., 1998). ArpA binds A-factor, an acyl-γ-butyrolactone (GBL), with high affinity. In the absence of A-factor, ArpA binds to specific sequences in the promoters of target genes, and prevents their expression. When ArpA binds A-factor, it loses its DNA-binding activity, and the target genes are expressed.
[0012]A-factor-like GBLs are a widespread family of molecules in streptomycetes, which accumulate outside the cells in culture; they appear to be freely exchanged between the cytoplasm and the medium. Only at high cell density does the concentration outside, and therefore inside, the cells become sufficient to cause detachment of cognate binding proteins (such as ArpA) from promoters. The result of this is that the target genes become active only at high cell density. Moreover, at least some of these target genes regulate sporulation and/or antibiotic production, processes which occur only in dense cultures.
[0013]The inventors have also discovered that the deduced amino acid sequence of the mmfL gene is very significantly similar to proteins which confer GBL production in other Streptomyces spp. (FIG. 3). It can be seen in FIG. 1 that the mmfL gene is located between the two repressor-encoding genes mmyR and mmfR, along with mmfP and mmfH.
[0014]Further, the inventors present experimental results, based on the insertion of a marker gene into the mmyG and mmfH genes, which show that mmyG and mmfH are selectively expressed at high cell density (FIG. 4). The gene chosen was xylE from a plasmid of pseudomonads (Bruton et al, 1991) The xylE gene product is the enzyme catechol oxygenase, which may be detected by colony staining (Ingram et al., 1989).
[0015]From sequence analysis, it newly appears that the mmyT, mmyO and mmyG genes are transcribed from a common promoter, within the non-coding region between mmfR and mmyT. Thus, the inventors suggest that all three genes of the mmyTOG region are selectively expressed at high cell density. Similarly, it newly appears that mmfL, mmfH and mmfP are transcribed from a common promoter, within the non-coding region between mmfL and mmfR, and that this promoter is similarly regulated.
[0016]Similar regulation of expression was obtained when xylE was inserted into the mmy . . . XCAPK transcription unit (FIGS. 4 and 5), indicating that the promoter for this region is similarly regulated.
[0017]Based on their studies, the inventors now provide a model for the regulation of methylenomycin production in Streptomyces. The products of the mmyR and/or mmfR genes, ArpA homologues, bind to the promoter(s) of gene(s) presumed to encode methylenomycin biosynthetic enzymes or positive regulators of methylenomycin biosynthesis, thereby preventing methylenomycin production. MmfL, the product of the mmfL gene, directs the synthesis of a GBL, which binds to the products of the mmyR and/or mmfL genes. At sufficiently high cell density (and hence sufficiently high mmfL-specified GBL concentration in the medium), this latter binding is sufficient to prevent binding of the mmyR and/or mmfR gene products to the promoter(s) whose activation is necessary for methylenomycin production. Included among these are the promoters of mmyTOG, mmfLHP and mmy . . . XCAPK. It is further deduced that the induction of the mmfLHP promoter forms a "gearing" system, amplifying the production of GBL, and "committing" the cells to uninhibited expression from the mmyTOG and mmy . . . XCAPK promoters.
[0018]As a result of further experiments, it is further suggested that the product of another gene, mmyB, is also involved in this commitment (see Example 4).
[0019]The inventors now predict from this model that disruption of the mmyR and/or mmfR genes will cause increased methylenomycin production, because loss of repressors releases target promoters from repression. Observations have confirmed this prediction and show that none of the mmyR, mmfP and mmfH genes is a positive regulator; that none of them encodes an essential biosynthetic enzyme for methylenomycin biosynthesis; and that mmyR acts negatively.
[0020]As a result of further experiments, the inventors also predict that an additional gene, mmyB, is also involved in the operation of the regulatory scheme. Observations reported in Example 4 have supported this prediction.
[0021]The inventors also believe that the presence of an extra copy of mmfL would cause increased methylenomycin production, since this should lead to increased GBL synthesis and hence more efficient lifting of repression. Observations have confirmed this prediction also.
[0022]The sequencing of the mmfL gene shows that it contains an unusual feature, namely the presence of a TTA codon (position shown in FIG. 1). TTA (=UUA in mRNA) is one of six codons which encode leucine. However, from the fact that there are fully viable mutant strains of Streptomyces which lack the ability to translate this codon, it is known that the TTA codon is not used in any essential genes of Streptomyces spp. Such mutants are defective in the bldA gene which directly encodes the transfer RNA responsible for recognising the UUA codon (Leskiw et al., 1991). Translation of mmfL mRNA into MmfL protein would therefore be severely impaired in a bldA mutant. Observations reported in Example 4 have strongly supported this prediction.
[0023]The present inventors also predict that there would be a failure of GBL production in a bldA mutant, which would lead to non-production of methylenomycin, since their model indicates that mmfL confers production of a GBL needed to relieve repression of methylenomycin synthesis. Observations reported in Example 4 have strongly supported this prediction.
[0024]Experiments are described herein in which an appropriately orientated foreign gene encoding an easily detectable enzyme (xylE) was inserted into different transcription units in the methylenomycin gene cluster.
[0025]Expression of xylE was detected at high levels in, for example, bldA+ strains carrying mmyG::xylE, mmfH::xylE or mmy . . . XCAPK::xylE fusions, but was undetectable in bldA mutants carrying the same fusions, confirming the prediction of the model.
[0026]The pattern of expression of catechol oxygenase in bldA+ strains carrying mmyG::xylE, mmy . . . XCAPK::xylE or mmfH::xylE fusions was that early in growth there was no detectable activity, whereas later in growth the specific activity rose very sharply (FIG. 4), demonstrating that the promoter driving xylE expression is very specifically regulated.
[0027]For comparison when xylE was fused to the redX transcription unit for production of undecylprodigiosin, a different antibiotic, in the chromosome of S. coelicolor, much lower specific activity was obtained, albeit in different growth conditions (Guthrie and Chater, 1990). Thus the promoter driving mmyG::xylE expression is very strong, by comparison to those of genes for other Streptomyces antibiotics. Similarly strong expression is indicated for the promoters driving expression of mmyK and mmfH.
[0028]In continuing sequencing of the methylenomycin genes one other target TTA sequence for bldA action has been discovered, indicating that the TTA codon in mmfL may not be the sole reason for bldA-dependence of methylenomycin production. Observations have verified this prediction (see Example 4b).
[0029]From their model, the inventors teach that the insertion of a nucleic acid of interest (e.g. a gene or genes) into the mmyTOG region, in the correct orientation, and in the presence of the mmyR-mmf-mmyTOG region left of the insertion site, provides self-regulating, strong expression of the nucleic acid of interest specifically late in culture, to give a high level of expression only in conditions when the main growth phase has been completed (i.e. at high biomass and high mycelium density). This has four main advantages for the expression of the nucleic acid of interest: (1) reduced or no expression earlier in growth, avoiding toxic effects of some gene products on growth; (2) there is no requirement for an exogenously added inducer, avoiding various constraints on the culture medium or problems of the cost of adding inducers to large fermenters or of removing them from the desired end product; (3) the methylenomycin cluster, naturally present on a highly transmissible plasmid, is likely to have evolved to permit properly regulated expression in diverse Streptomyces hosts (see above), which is important commercially because virtually every antibiotic or other Streptomyces product made commercially involves a different strain; and (4) the expression is driven by a strong promoter, leading to high yield of the desired end product. Further experiments appear to indicate that the host strain used for expression should contain those mmy genes to the right of mmr, or at least the mmyB gene.
[0030]The inventors similarly teach that similar results could be obtained with the nucleic acid of interest inserted in the appropriate orientation into the mmy . . . XCAPK region, in the presence of the mmy . . . XCAPK region to the right of the insertion site and in the presence of the mmy . . . XCAPK promoter, or with the nucleic acid of interest inserted in the appropriate orientation into the mmfLHP region, in the presence of the mmfLHP region to the right of the insertion site and in the presence of the mmfLHP promoter.
[0031]Similar results may also be obtained with the nucleic acid of interest inserted in the appropriate orientation into the mmyBQE region, in the presence of the mmyBQE region to the right of the insertion site and in the presence of the mmyBQE promoter, or with the nucleic acid of interest inserted in the appropriate orientation into the mmyYF region, in the presence of the mmyYF region to the left of the insertion site and in the presence of the mmyYF promoter.
[0032]Furthermore, it is believed that certain regions of the 9.5 kb stretch which has been investigated are of greater importance than others. In particular, the model teaches that interplay between the products of the mmfR, mmyR, mmfL and mmyB genes and the promoters of the mmyTOG, mmy . . . XCAPK and mmfLHP regions is key to the regulation of methylenomycin production. Consequently, it is taught that the combination of the nucleic acid of interest and minimal regulatory portions which include an mmfR gene and/or an mmyR gene; an mmfL gene; an mmyB gene; and an mmyTOG promoter and/or an mmy . . . XCAPK promoter and/or an mmfLHP promoter and/or an mmyBQE promoter and/or an mmyYF promoter will also lead to increased expression of the nucleic acid of interest at higher cell density relative to lower cell density.
[0033]However, it is also contemplated that the mmfP and mmfH genes may be of importance in regulation of methylenomycin production, for example in some conditions their products may modify the structure of the GBL whose production is conferred by the mmfL gene, resulting in changes in the details of interactions between the GBL and the mmyR and/or mmfR gene products. Consequently these genes may also be present in the regulatory portion.
[0034]Moreover, having found that three different promoters of methylenomycin biosynthetic and positive regulatory genes are regulated in the same way, the inventors expect that other promoters of methylenomycin biosynthetic genes will also be similarly regulated.
[0035]In a first aspect, therefore, the present invention provides an expression cassette for the expression of a nucleic acid of interest, the expression cassette including: [0036]a regulatory portion or portions including: [0037]a first regulatory element which includes either an mmyR gene encoding an MmyR polypeptide, or an mmfR gene encoding an MmfR polypeptide, or both; [0038]a second regulatory element which includes an mmfL gene encoding an MmfL polypeptide; and [0039]a promoter (the "repressible promoter"), the function of which is repressed by the expression product of the first regulatory element, that repression being alleviated or removed by a product, the production of which is conferred by the MmfL polypeptide, and [0040]the nucleic acid of interest, in operative association with said promoter.
[0041]Such a construct represents the minimal expression cassette which may be predicted by the above model to cause expression of the nucleic acid of interest in Streptomyces at high cell density.
[0042]Preferably, the expression cassette is capable of expressing the nucleic acid of interest at increased levels in stationary phase cultures of Streptomyces compared to early exponential phase cultures.
[0043]Preferably the regulatory elements, promoter and nucleic acid of interest are provided on a single expression cassette, but it is contemplated that they may be provided separately, for example on two vectors which may be co-introduced into a desired host, or that one or more of the regulatory elements and promoter may be provided by the SCP1 plasmid.
[0044]Accordingly in this first aspect, the present invention also provides a set of nucleic acids for the expression of a nucleic acid of interest, the set of nucleic acids together including: [0045]a regulatory portion or portions including: [0046]a first regulatory element which includes either an mmyR gene encoding an MmyR polypeptide, or an mmfR gene encoding an MmfR polypeptide, or both; [0047]a second regulatory element which includes an mmfL gene encoding an MmfL polypeptide; [0048]a promoter (the "repressible promoter"), the function of which is repressed by the expression product of the first regulatory element, that repression being alleviated or removed by a product, the production of which is conferred by the MmfL polypeptide, and [0049]the nucleic acid of interest, in operative association with said promoter.
[0050]Preferably the set is an isolated set of nucleic acids.
[0051]Preferably both an mmyR gene and an mmfR gene will be provided in the regulatory portion(s) of this aspect.
[0052]Preferably an mmyB gene encoding an MmyB polypeptide will be provided within, or in addition to, the expression cassette or set of nucleic acids, as a mediator of the regulatory effects of the regulatory portion. In a preferred embodiment, the mmyB gene is incorporated into a third regulatory element of the regulatory portion(s). Preferably some or all of other mmy genes to the right of the mm gene (as shown in FIG. 1e) are also provided.
[0053]The repression of the function of the repressible promoter by the expression product of the first regulatory element may be direct repression by the expression product, or it may arise from the absence of an activator which is itself repressed by the first regulatory element.
[0054]Preferably the regulatory portion(s) comprise at least a portion of the 9.5 kb newly sequenced stretch of nucleic acid as shown in FIG. 7, or a variant thereof.
[0055]Preferably, the mmyR gene encodes an MmyR polypeptide having the amino acid sequence of FIG. 8a (optionally excluding the first 6 listed amino acids). More preferably it comprises residues 1407 to 796 of FIG. 7, lower strand (optionally excluding residues 1407 to 1390).
[0056]Preferably, the first gene encodes an MmfR polypeptide having the amino acid sequence of FIG. 8E. More preferably it comprises residues 4807 to 5451 of FIG. 7, upper strand.
[0057]Preferably, the first regulatory element also includes a promoter operatively linked to the mmyR or mmfR gene. In embodiments where both genes are present, they are preferably operatively linked to respective promoters. Still more preferably, the promoter to which the mmyR gene is linked is an mmyR promoter and/or the promoter to which the mmfR gene is linked is an mmfR promoter. Even more preferably the promoter to which the mmyR gene is linked comprises some or all of residues 1557 to 1390 of FIG. 7, lower strand (optionally excluding residues 1409 to 1390) and/or the promoter to which the mmfR gene is linked comprises some or all of residues 4613 to 4806 of FIG. 7, upper strand.
[0058]Preferably, the mmfL gene encodes an MmfL polypeptide having the amino acid sequence of FIG. 8d. More preferably, it comprises residues 4612 to 3551 of FIG. 7, lower strand.
[0059]Preferably, the second regulatory element also includes a promoter operatively linked to the mmfL gene. Still more preferably, the promoter to which the mmfL gene is linked is an mmfL promoter. Even more preferably the promoter to which the mmfL gene is linked comprises some or all of residues 4806 to 4613 of FIG. 7, lower strand.
[0060]It will be observed that the preferred promoters for mmfL and mmfR both comprise some or all of residues 4806 to 4613 of FIG. 7. This region is thought to include a bi-directional promoter for both these genes. Preferably the promoter(s) for mmfL and/or mmfR include a palindromic sequence having a high degree of sequence identity or complete sequence identity with a palindromic sequence having the half-site 5'-GG(T/C)CGGT(A/T)(T/C)G(T/G)-3', which is the consensus sequence for binding of DNA by the ArpA protein. In this context high sequence identity preferably represents sequence identity at seven or more, more preferably 8 or more, 9 or more, or 10 or more corresponding positions within the half-site. More preferably the palindromic sequence has the half-site 5'-GGAAGGTATTA-3' (or a variant thereof). In a particularly preferred embodiment, the regulatory portion comprises residues 3551 to 5451 of FIG. 7, upper strand (or a variant thereof).
[0061]Preferably the repressible promoter is a promoter of a methylenomycin biosynthetic or regulatory gene.
[0062]More preferably the repressible promoter comprises an mmyTOG promoter or an mmy . . . XCAPK promoter or an mmfLHP promoter or an mmyBQE promoter or an mmyYF promoter.
[0063]However, it is also thought that other promoters of methylenomycin biosynthetic genes may be regulated in the same way as mmyTOG, mmy . . . XCAPK and mmfLHP. Accordingly, the repressible promoter may comprise a promoter of any other methylenomycin biosynthetic gene which is regulated by the mmyR and/or mmfR genes and the mmfL gene, typically mediated by the mmyB gene.
[0064]Preferably the mmyTOG promoter comprises some or all of residues 5452 to 5675 of FIG. 7; upper strand.
[0065]Preferably the mmfLHP promoter comprises some or all of residues 4613 to 4806 of FIG. 7, lower strand.
[0066]The mmy . . . XCAPK, mmyBQE and mmyYF promoters remain to be accurately located. However, this may be accomplished routinely by sequencing and sequence analysis of, for example, the restriction fragments A4.2 and A3.13 (Chater and Bruton, 1983), which make available different parts of the mmy . . . XCAPK cluster, or by analysis of EMBL accession number AJ276673. It is thought that the mmy . . . XCAPK promoter comprises some or all of residues 15404 to 15977 of EMBL AJ276673 and that the mmyBQE promoter comprises some or all of residues 18892 to 19123 of EMBL AJ276673, or that the mmyBQEDXCAPK genes are all transcribed from a single promoter which comprises some or all of residues 18892 to 19123 of EMBL AJ276673. It is thought that the mmyYF promoter comprises some or all of residues 18892 to 19123 of EMBL AJ276673.
[0067]Apart from the repressible promoter, the nucleic acid of interest is preferably not additionally in operative association with any exogenous promoter, i.e. any promoter which is not derived from (or a variant of) a promoter present in the methylenomycin gene cluster. For example, in preparing the expression cassette (or set of nucleic acids) according to this aspect of the invention, the nucleic acid of interest is preferably brought into operative association with the repressible promoter in the functional absence of any promoter with which it may have previously been associated (for example a promoter of a cloning vector).
[0068]Preferably, the mmyB gene encodes an MmyB polypeptide having the deduced amino acid sequence shown for this gene in EMBL AJ276673. More preferably, it comprises the complement of residues 18032 to 18892 of the nucleic acid sequence shown in EMBL AJ276673.
[0069]Preferably, the third regulatory element also includes a promoter operatively linked to the mmyB gene. Still more preferably, the promoter to which the mmyB gene is linked is an mmyB promoter. Even more preferably the promoter to which the mmyB gene is linked comprises some or all of residues 18892 to 19123 of EMBL AJ276673.
[0070]Since it is thought that they may be of importance in directing expression to high cell density cultures in some fermentation conditions, an mmfP gene and/or an mmfH gene, preferably both, are preferably included in the regulatory portion(s). However in some embodiments, in which an mmyLHP promoter is operatively linked to the nucleic acid of interest, one or more of these genes may be replaced or disrupted by the nucleic acid of interest.
[0071]When an mmyTOG promoter is used, at least part of at least one of an mmyT, an mmyO and/or an mmyG gene is suitably present in the regulatory portion, in operative association with the mmyTOG promoter. However, there may in particular be a 3' (right hand end) truncation of the mmyTOG coding region.
[0072]When an mmfLHP promoter is used, at least part of at least one of an mmfH and/or an mmfP gene is suitably present in the regulatory portion, in operative association with the mmfLHP promoter. However, there may in particular be a 3' (left hand end) truncation of the mmfLHP coding region. Preferably an intact mmfL-gene is also in operative association with the same mmfLHP promoter.
[0073]When an mmy . . . XCAPK promoter is used, at least part of at least one of an mmyX, an mmyC, an mmyA, an mmyP, an mmyK gene, and/or an mmyD gene (mmyD is a co-transcribed gene located between mmyX and the putative mmy . . . XCAPK promoter) is suitably present in the regulatory portion, in operative association with the mmy . . . XCAPK promoter. However, there may in particular be a 3' (left hand end) truncation of the mmy . . . XCAPK coding region.
[0074]When an mmyBQE promoter is used, at least part of at least one of an mmyB, an mmyQ and/or an mmyE gene is suitably present in the regulatory portion, in operative association with the mmyBQE promoter. However, there may in particular be a 3' (left hand end) truncation of the mmyBQE coding region.
[0075]The same applies mutatis mutandis to the situation where an mmyBQEDXCAPK promoter is used.
[0076]When an mmyYF promoter is used, at least part of at least one of an mmyY and an mmyF gene is suitably present in the regulatory portion, in operative association with the mmyYF promoter. However, there may in particular be a 3' (right hand end) truncation of the mmyYF coding region.
[0077]The location and sequence of the individual genes with the mmyDXCAPK, mmyBQE (or mmyBQEDXCAPK) and mmyYF regions is given in EMBL AJ276673. The sequences of the corresponding promoters may be deduced by sequence analysis and/or routine experimentation on the basis of this sequence information (e.g. using DNase footprinting experiments).
[0078]Desirably, the nucleic acid of interest is inserted into the regulatory portion, preferably within an mmyT, mmyO or mmyG gene (when an mmyTOG promoter is used) or within an mmyD, mmyX, mmyC, mmyA, mmyP or mmyK gene (when an mmy . . . XCAPK promoter is used) or within an mmfH or mmfP gene (when an mmfLHP promoter is used) or within an mmyQ or mmyE gene (when an mmyBQE promoter is used) or within an mmyQ, mmyE, mmyD, mmyX, mmyC, mmyA, mmyP or mmyK gene (when an mmyBQEDXCAPK promoter is used) or within an mmyY or mmyF gene (when an mmyYF promoter is used).
[0079]The nucleic acid of interest may be inserted into the regulatory portion by means of homologous recombination (e.g. using a vector containing a fragment of the mmyTOG, mmy . . . XCAPK, mmyBQE, mmyBQEDXCAPK, mmyYF or mmfLHP coding region). Accordingly, the regulatory portion may contain an entire mmyTOG, mmy . . . XCAPK, mmyBQE, mmyBQEDXCAPK, mmyYF or mmfLHP coding region, which is interrupted by the nucleic acid of interest.
[0080]Preferably, the mmyT gene encodes an MmyT polypeptide having the amino acid sequence of FIG. 8f, or a variant thereof. More preferably, it comprises residues 5676 to 6401 of FIG. 7, upper strand.
[0081]Preferably, the mmyO gene encodes an MmyO polypeptide having the amino acid sequence of FIG. 8g. More preferably, it comprises residues 6432 to 7553 of FIG. 7, upper strand.
[0082]Preferably, the mmyG gene encodes an MmyG polypeptide having the amino acid sequence of FIG. 8h. More preferably, it comprises residues 7636 to 8817 of FIG. 7, upper strand.
[0083]Preferably, the mmfL gene encodes an MmfL polypeptide having the amino acid sequence of FIG. 8d. More preferably, it comprises residues 3551 to 4612 of FIG. 7, lower strand.
[0084]Preferably, the mmfH gene encodes an MmfH polypeptide having the amino acid sequence of FIG. 8C. More preferably, it comprises residues 3554 to 2352 of FIG. 7, lower strand.
[0085]Preferably, the mmfP gene encodes an MmfP polypeptide having the amino acid sequence of FIG. 8b. More preferably, it comprises residues 3554 to 1558 of FIG. 7, upper strand.
[0086]The same applies, mutatis mutandis, to the mmyDXCAPK, mmyBQE and mmyYF genes, based on the respective amino acid and nucleic acid sequences given in EMBL AJ276673.
[0087]Preferably the nucleic acid of interest is heterologous, i.e. having a sequence not present in or derived from the 9.5 kb stretch of newly sequenced DNA, more preferably not present in or derived from the methylenomycin biosynthetic gene cluster.
[0088]In one preferred embodiment, the regulatory portion(s) include(s) an mmfR gene and an mmfL gene, with the mmfL promoter as the repressible promoter (for example the nucleic acid may be inserted into the mmfH or mmfP gene, downstream of mmfL under the control of the mmfLHP promoter).
[0089]In a more preferred embodiment, the regulatory portion(s) include(s) an mmfR gene, an mmfL gene and an mmyR gene, with the mmyB promoter as the repressible promoter (for example the nucleic acid may be inserted into the mmyB, mmyQ or mmyE gene, under the control of the mmyB promoter).
[0090]In a still more preferred embodiment, the regulatory portion(s) include(s) an mmfR gene, an mmfL gene, and mmyR gene, with the mmyTOG promoter, the mmyDXCAPK or the mmyYF promoter as the repressible promoter, such regulatory portion(s) being suitable for use in the presence (either as a third regulatory portion or as another part of the expression system) of an mmyB gene.
[0091]In a highly preferred embodiment, the regulatory portion(s) may include nucleic acid having the sequence from residue 796 to a residue between 5676 and 8817 (inclusive) of FIG. 7 (or a variant thereof). When double stranded, this nucleic acid contains mmyR, mmfP, mmfH, mmfL and mmfR genes and intergenic regions between those genes and at least a region containing an mmyTOG promoter (i.e. the region upstream of mmyT), and optionally some or all of an mmyTOG coding region. The nucleic acid of interest may then be inserted into or downstream of the mmyTOG region (or part thereof) in operative association with the mmyTOG promoter.
[0092]In another highly preferred embodiment, the regulatory portion(s) may include nucleic acid having the sequence from residue 796 to residue 5451 (inclusive) of FIG. 7 (or a variant thereof). When double stranded, this nucleic acid contains mmyR, mmfP, mmfH, mmfL and mmfR genes and intergenic regions between those genes. The nucleic acid of interest may then be inserted into the mmfHP region in operative association with the mmfLHP promoter.
[0093]It is contemplated that elements contributing to promoter activity (e.g. enhancer elements) may lie outside the non-coding intergenic regions specified above. Accordingly, it is particularly preferred that the intergenic promoter regions are located in their natural immediate context, i.e. between genes (or parts of genes) which normally flank them. Accordingly embodiments using a cassette containing an intact mmyR-mmf-mmyTOG region, optionally with a disruption of or 3' (right-hand end) truncation of the mmyTOG region, are particularly preferred.
[0094]In a second aspect, the present invention provides a vector comprising an expression cassette according to the first aspect of the invention. Further it provides a set of vectors comprising a set of nucleic acids according to the first aspect of the invention.
[0095]Suitable vectors comprising nucleic acid for introduction into bacteria can be chosen or constructed, containing appropriate additional regulatory elements if necessary, including additional promoters, terminator fragments, enhancer elements, marker genes and other elements as appropriate. Vectors may be plasmids, viral eg "phage", or "phagemid", as appropriate. For further details see, for example, Sambrook et al, (1989). Many known techniques and protocols for manipulation of nucleic acid, for example in preparation of nucleic acid constructs, mutagenesis, sequencing, introduction of DNA into cells and gene expression, and analysis of proteins, are described in detail in Ausubel et al. (1992). Many aspects of the employment of these techniques in the context of Streptomyces spp. are described in detail in Hopwood et al (1985) and Kieser et al (2000). The disclosures of Sambrook et al, Ausubel et al, Hopwood et al and Kieser et al are all incorporated herein by reference for these and all other purposes.
[0096]In a third aspect, the present invention provides an expression system comprising an expression cassette or set of nucleic acids according to the first aspect of the invention and an expression system comprising a vector according to the second aspect of the invention.
[0097]Preferably the expression system is a host cell, although cell-free expression systems are also contemplated. Preferably the host cell is a bacterium, more preferably an actinomycete, further preferably a streptomycete. In particular, it has been shown (see Examples) that the invention can be applied successfully in streptomycete strains other the S. coelicolor, as expected on the basis of the transmissibility of the plasmid SCP1.
[0098]Preferably the expression system (usually a native or genetically modified host cell) contains an mmyB gene, more preferably as an additional part of the vector system used to introduce the expression cassette/set f nucleic acids. However, it is also contemplated that the mmyB gene may be present e.g. as part of the host cell genome and/or on a plasmid, e.g. SCP1 or pSV1 also present within the expression system. This is a less preferred embodiment as other methylenomycin gene promoters may sequester mmyB gene product, reducing its effectiveness in mediating expression from the expression cassette/set of nucleic acids.
[0099]In one preferred embodiment, the expression system lacks the ability to translate the codon TTA (UUA in mRNA), and the expression cassette, set of nucleic acids or vector(s) lacks TTA codons, and/or has been modified to eliminate one or more (preferably all) naturally occurring TTA codons. For example the expression system is preferably a cell of a blDA mutant strain of Streptomyces and the expression cassette preferably contains a variant of the mmfL gene in which the naturally occurring TTA codon has been altered (e.g. by site-directed mutagenesis) into another leucine-encoding codon. Preferably the mmyB gene included in this system (whether as part of an expression cassette or part of the host cell genome or on a plasmid also present in the system) has also been similarly altered so that its TTA codon has been changed to another leucine-encoding codon (see Example 10). This provides the advantage, particularly in Streptomyces spp., that expression of the nucleic acid of interest can be achieved with reduced expression of other products (for example antibiotics) which would otherwise also be expressed at high cell density via bldA-dependent mechanisms; i.e. this system provides a clean background for expression of the nucleic acid of interest. One or more TTA codons are present in the biosynthetic gene clusters for most streptomycete antibiotics, particularly in pathway-specific regulatory genes. TTA codons should not be present in genes whose expression is being carried out in a bldA mutant host. In some circumstances it may be necessary to use site-directed mutagenesis to convert a TTA codon to another leucine codon.
[0100]In another preferred embodiment the number of repressible promoters present in the expression system is limited. This may reduce expression-limiting sequestration of mmyB gene product by promoters other than the repressible promoter controlling expression of the nucleic acid of interest. Such limitation may involve the absence from the expression system of the SCP1 plasmid and/or the pSV1 plasmid (though in such cases the mmyB gene is preferably otherwise present in the expression system).
[0101]Additionally or alternatively, the expression cassette/set of nucleic acids may lack repressible promoters other than the promoter controlling expression of the nucleic acid of interest (however, promoters controlling expression of other genes of the expression cassette/set of nucleic acids may for this purpose be regarded as not being repressible promoters, e.g. the mmyTOG promoter may control the expression of the nucleic acid of interest and the mmfL and mmyB genes may be controlled by their native promoters). Lacking may be relative (i.e. there are fewer repressible promoters than in the native methylenomyin cluster), substantial or complete.
[0102]The introduction of the expression cassette, set of nucleic acids or vector(s) into a host cell, which may (particularly for in vitro introduction) be generally referred to without limitation as "transformation", may employ any available technique. For bacterial cells, suitable techniques may include calcium chloride transformation, polyethyleneglycol assisted transformation, electroporation, conjugation and transfection or transduction using bacteriophages.
[0103]In a fourth aspect, the present invention provides a method of expressing a nucleic acid of interest, the method comprising providing a host cell (or other expression system) according to preferred embodiments of the third aspect and culturing the host cell, so as to express the nucleic acid of interest.
[0104]Preferably the nucleic acid of interest is expressed substantially only when the host cell culture reaches high cell density, more preferably at or close to the stationary phase of host cell culture. Cell cultures at or close to stationary phase may have ODb50 values in the range of 1-20.
[0105]Known methods of culturing cells are well known in the art, for example from Sambrook et al (1989), Ausubel et al (1992), and (in particular for Streptomyces spp.) Hopwood et al (1985) and Kieser et al (2000).
[0106]In a fifth aspect, the present invention provides a method of expressing a nucleic acid of interest, the method comprising: [0107]providing in an expression system a regulatory portion or portions as defined in the first aspect; [0108]providing in the expression system the nucleic acid of interest; [0109]operatively associating the nucleic acid of interest with the repressible promoter of the regulatory portion(s); and [0110]expressing the nucleic acid of interest in the expression system.
[0111]The steps of the method need not be performed in the order recited. In particular, the operative association may occur prior to introduction regulatory portion(s) and nucleic acid of interest into the expression system.
[0112]The preferred features specified for the regulatory portion(s) in the context of the first aspect of the invention may also be present in the regulatory portion(s) used in this aspect. Preferably the expression system is a cell, more preferably a bacterium, further preferably an actinomycete and most preferably a streptomycete. Preferably the expression system contains an mmyB gene which may be introduced together with the regulatory cassette. The cell is preferably cultured for expression of the nucleic acid of interest.
[0113]The nucleic acid of interest may be brought into operative association with the repressible promoter in a variety of ways. For example, the nucleic acid of interest may be inserted into a nucleic acid molecule which contains the repressible promoter, downstream of the repressible promoter (FIGS. 6c and 6d). In a preferred example, the repressible promoter is an mmyTOG promoter and the insertion site is within an mmyTOG region.
[0114]Alternatively, the regulatory portion may be inserted into nucleic acid containing the nucleic acid of interest, for example by homologous recombination (FIG. 6b). Thus a fragment from the 5' end of the nucleic acid of interest may be included downstream of and in operative association with the repressible promoter to permit homologous recombination of the regulatory portion into the nucleic acid of interest.
[0115]This method may be advantageously used in a bldA mutant Streptomyces or other actinomycete host cell, with a regulatory portion or portions (and preferably mmyB gene) lacking TTA codons, to regulate the expression of a nucleic acid of interest which is native to the host cell and which preferably confers production of an antibiotic. This embodiment has the advantage that other antibiotics encoded by the host cell will generally not be expressed, since the pathway-specific regulatory genes for production of such other antibiotics in Streptomyces typically include a TTA codon. For example, in one preferred expression system, S. coelicolor A(3)2, major pathway-specific regulatory genes of each of two known chromosomally located antibiotic pathways contain TTA codons, and a bldA mutant therefore makes neither of these antibiotics in typical culture media (Fernandez-Moreno et al. 1991, White and Bibb 1997).
[0116]The expression products of the nucleic acids of interest of the fourth and fifth aspects may be collected and purified. This may be achieved by conventional methods. See for example McDaniel et al. (1993).
[0117]Where the nucleic acid of interest is for example a biosynthetic gene cluster, both the end product of the biosynthesis and the biosynthetic enzymes themselves may be regarded to be the expression product, but more usually the end product will be regarded to be the expression product.
[0118]In a sixth aspect, therefore, the present invention provides an expression product produced according to the method of either of the fourth or fifth aspects of the invention.
[0119]The nucleic acid of interest may be any nucleic acid. Preferred nucleic acids are genes, the expression of which is desired, or gene clusters, for example which encode the enzymes necessary for the biosynthesis of e.g. antibiotics. Gene clusters may have a plurality of genes within the same transcriptional promoter, so as to allow expression of all genes of the cluster from the repressible promoter. Alternatively, the nucleic acid of interest may be an unknown nucleic acid which it is desired to investigate, for example nucleic acid derived from a sample e.g. of soil.
[0120]In a seventh aspect, the present invention provides a nucleic acid molecule comprising an mmyR and/or an mmfR gene, an mmfL gene, and a repressible promoter, all as defined in the first aspect, wherein the molecule is capable of regulating the expression of a nucleic acid of interest when that nucleic acid is arranged in operative association with the repressible promoter.
[0121]The same preferred and optional features apply to this aspect as they apply to the first aspect.
[0122]Preferably the molecule of this aspect is other than pIJ519, as disclosed in Chater and Bruton (1985) and/or does not consist of or include the 350 kb SCP1 plasmid of Streptomyces coelicolor and/or the pSV1 plasmid of Streptomyces violaceoruber. However, such plasmids may be used in conjunction with the molecule of this aspect (e.g. to supply an mmyB gene).
[0123]Preferably, the molecule consists essentially of an approximately 4.8 to 8 kb stretch of DNA including mmyR, mmfP, mmfH, mmfL and mmfR genes and at least a portion of at least one of an mmyT gene, an mmyO gene and an mmyG gene. More preferably, the molecule includes the entire mmyT and mmyO genes and at least a portion of the mmyG gene.
[0124]Preferably the molecule consists essentially of the nucleic acid having the sequence from residue 796 to a residue between 5676 and 8817 (more preferably between 7636 and 8817) of FIG. 7.
[0125]In an alternative embodiment, the molecule consists essentially of a stretch of nucleic acid including mmyR, mmfP, mmfH, mmfL and mmfR genes in combination with a stretch of nucleic acid including an mmy . . . XCAPK promoter and at least a portion of at least one of an mmyD gene, an mmyX gene, an mmyC gene, an mmyA gene, an mmyP gene, an mmyK gene.
[0126]In an alternative embodiment, the molecule consists essentially of an approximately 5-kb stretch of DNA including mmyR, mmfP, mmfH, mmfL and mmfR genes.
[0127]In addition to the defined stretches of DNA, the molecule preferably comprises an mmyB gene.
[0128]In an eighth aspect, the present invention provides a nucleic acid molecule consisting essentially of one or more of an mmyB gene in which a naturally occurring TTA codon has been changed into another (preferably leucine encoding) codon, an mmyR gene, an mmfP gene, an mmfH gene, an mmfL gene (preferably in which a naturally occurring TTA codon has been changed into another (preferably leucine encoding) codon), an mmfR gene, an mmyT gene, an mmyO gene, and an mmyG gene, optionally with a respective upstream region or respective upstream regions. Where present, the upstream region(s) may comprise promoters (preferably as previously defined) for the genes. Where two or more genes are present, an upstream region for one or more of those genes may be provided in an intergenic region.
[0129]The genes may be as previously defined.
[0130]In a ninth aspect, the present invention provides the use of one or more nucleic acid molecules as defined in any one of the seventh or eighth aspects, in or for the regulation of expression of a nucleic acid of interest in an expression system.
[0131]In a tenth aspect, the present invention provides a polypeptide encodable by one of the following genes: an mmyR gene, an mmfP gene, an mmfH gene, an mmfL gene, an mmfR gene, an mmyT gene, an mmyO gene and an mmyG gene. Preferably the polypeptide is substantially isolated from other proteins with which it is naturally associated.
[0132]The polypeptide preferably has an amino acid sequence as shown in one of FIGS. 8a to 8h. However, this aspect also provides polypeptides which are variants of those amino acid sequences.
[0133]In an eleventh aspect, the present invention provides a vector including a nucleic acid according to the seventh or eighth aspect. In embodiments of the seventh and eighth aspects in which the nucleic acid lacks a promoter or promoters for the gene or genes it contains, the vector preferably includes a promoter in operative association with that gene or those genes.
[0134]In a twelfth aspect, the present invention provides an expression system containing one or more nucleic acids according to the seventh or eighth aspects and an expression system containing a vector according to the eleventh aspect.
[0135]Preferably the expression system is a cell. Where it is desired merely to express the polypeptide encoded by the nucleic acid, rather than for example to regulate the expression of another nucleic acid of interest, any appropriate cell may be used (e.g. a standard E. coli overexpression system). See for example Sambrook et al (1989) and Ausubel et al (1992). Otherwise, bacterial, actinomycete and streptomycete cells are preferred as previously indicated.
[0136]In a thirteenth aspect, the present invention provides a method of producing a polypeptide according to the tenth aspect, the method comprising producing the polypeptide in an expression system according to the twelfth aspect.
[0137]The polypeptide may be purified from the expression system by conventional methods.
[0138]References herein to genes, coding regions and nucleic acids are not to be interpreted as being restricted to genes, coding regions and nucleic acids having the specific nucleic acid sequences disclosed herein or in EMBL AJ276673. Rather, genes, coding regions and nucleic acids having variants of those sequences are also included. Genes, coding regions and nucleic acids having such specific sequences are preferred embodiments. Thus, for example, a reference to "an mmfR gene" is not to be interpreted as being restricted to a gene having the sequence from residue 4807 to residue 5451 of FIG. 7, but also includes variants.
[0139]Similarly, references herein to polypeptides are not to be interpreted as being restricted to polypeptides having the specific amino acid sequences disclosed herein or in EMBL AJ276673. Rather, polypeptides having variants of those sequences are also included. Polypeptides having such specific sequences are preferred embodiments. Thus, for example, a reference to "an MmfR polypeptide" is not to be interpreted as being restricted to a polypeptide having the amino acid sequence shown in FIG. 8e, but also includes variants.
[0140]References herein to promoters are not to be interpreted as being restricted to nucleic acids having the sequence of all or part of a specific intergenic region disclosed herein or in EMBL AJ276673. Again, promoters having variants of those intergenic sequences are also included and the specific intergenic sequences (or parts thereof) are preferred embodiments. Thus, for example, a reference to "an mmyTOG promoter" is not to be interpreted as being restricted to the specific mmyTOG promoter disclosed herein (i.e. a nucleic acid having all or part of the sequence from residues 5452 to 5675 of FIG. 7, upper strand), but also includes variants.
[0141]In all cases, where a preferred embodiment of a gene, nucleic acid, polypeptide or promoter is defined by reference to a specific sequence, the invention in its broader sense is intended to include embodiments having variants of that specific sequence.
[0142]The term "variant" as used herein in relation to a particular nucleic acid (the reference nucleic acid) denotes: any nucleic acid having a sequence which is different from that of the reference nucleic acid, but which is its complement or which shows significant nucleic acid sequence identity with, or hybridisation under stringent conditions to, the reference nucleic acid or its complement or a fragment of the reference nucleic acid or its complement; or any nucleic acid which encodes an amino acid sequence having significant amino acid sequence identity with the amino acid sequence encoded by the reference nucleic acid, or a fragment of that nucleic acid. The term "variant" also refers to nucleic acids which differ from each other due only to the degeneracy of the genetic code, and which therefore encode identical deduced amino acid sequences.
[0143]The term "variant" as used herein in relation to a particular polypeptide (the reference polypeptide) denotes: any polypeptide having an amino acid sequence which is different from, but which shows significant amino acid sequence identity with, the amino acid sequence of the reference polypeptide, or a fragment of that polypeptide.
[0144]Unless otherwise specified, significant amino acid sequence identity is preferably at least 80%, more preferably 85%, 90% or 95%, still more preferably 98% or 99% and/or significant nucleic acid sequence identity is preferably at least 50%, more preferably 60%, 70%, 80% or 90%, still more preferably 95%, 98% or 99%.
[0145]Significant amino acid sequence identity is preferably shown between the variant polypeptide (or a portion thereof) and a fragment of at least 10 amino acids of the reference polypeptide, more preferably a fragment of a least 20, 30 or 40 amino acids, still more preferably a fragment of 60, 80 or 100 amino acids, more preferably the entire reference polypeptide.
[0146]Significant nucleic acid sequence identity is preferably shown between the variant nucleic acid (or a portion thereof) and a fragment of at least 30 residues of the reference nucleic acid, more preferably a fragment of a least 60, 90 or 120 residues, still more preferably a fragment of 180, 240 or 300 residues, more preferably the entire reference nucleic acid.
[0147]In relation to variants of the specific mmyR gene disclosed herein, or of its product, MmyR, significant amino acid sequence identity is preferably shown with residues 40 to 49 of FIG. 8a, more preferably residues 38 to 49, more preferably residues 38 to 56, more preferably residues 38 to 59, still more preferably residues 21 to 59, still more preferably residues 3 to 59 and/or significant nucleic acid sequence identity is preferably shown with corresponding portions of FIG. 7 which encode the above amino acid residues.
[0148]In relation to variants of the specific mmfR gene disclosed herein, or of its product, MmfR, significant amino acid sequence identity is preferably shown with residues 61 to 70 of FIG. 8E, more preferably residues 59 to 70, more preferably residues 59 to 77, more preferably residues 59 to 80, still more preferably residues 42 to 80, still more preferably residues 24 to 80 and/or significant nucleic acid sequence identity is preferably shown with corresponding portions of FIG. 7 which encode the above amino acid residues.
[0149]In relation to variants of the specific mmfL gene disclosed herein, or of its product, MmfL, significant amino acid sequence identity is preferably shown with residues 77 to 87 and/or residues 240 to 255 of FIG. 8d, more preferably residues 77 to 95 and/or residues 231 to 255, more preferably residues 77 to 107 and/or residues 223 to 255 and/or significant nucleic acid sequence identity is preferably shown with corresponding portions of FIG. 7 which encode the above amino acid residues.
[0150]Percent (%) amino acid sequence identity" is defined as the percentage of amino acid residues in a candidate sequence that are identical with the amino acid residues in the sequence with which it is being compared, after aligning the sequences and introducing gaps, if necessary, to achieve the maximum percent sequence identity, and not considering any conservative substitutions as part of the sequence identity. The % identity values used herein are generated by WU-BLAST-2 which was obtained from Altschul et al. (1996); http://blast.wustl/edu/blast/README.html. WU-BLAST-2 uses several search parameters, most of which are set to the default values. The adjustable parameters are set with the following values: overlap span=1, overlap fraction=0.125, word threshold (T)=11. The HSPS and HSPS2 parameters are dynamic values and are established by the program itself depending upon the composition of the particular sequence and composition of the particular database against which the sequence of interest is being searched; however, the values may be adjusted to increase sensitivity. A % amino acid sequence identity value is determined by the number of matching identical residues divided by the total number of residues of the "longer" sequence in the aligned region, multiplied by 100. The "longer" sequence is the one having the most actual residues in the aligned region (gaps introduced by WU-BLAST-2 to maximize the alignment score are ignored).
[0151]Percent (%) nucleic acid sequence identity" is defined as the percentage of nucleotide residues in a candidate sequence that are identical with the nucleotide residues in the sequence under comparison. The identity values used herein were generated by the BLASTN module of WU BLAST-2 set to the default parameters, with overlap span and overlap fraction set to 1 and 0.125, respectively.
[0152]In relation to variants of the promoters used in the present inventions nucleic acid sequence identity is preferably assessed over a sequence of at least 30 residues, more preferably 40 or 50 residues, still more preferably 60 residues. Thus, for example, preferred variants of the embodied mmyTOG promoter may have sequences which show 80% (or more) sequence identity over a 30 (or more) residue sequence within residues 5452 to 5675 of FIG. 7, upper strand.
[0153]Stringent conditions" or "high stringency conditions", as defined herein, may be identified by those that: (1) employ low ionic strength and high temperature for washing, for example 0.015 M sodium chloride/0.0015 M sodium citrate/0.1% sodium dodecyl sulfate at 50° C.; (2) employ during hybridization a denaturing agent, such as formamide, for example, 50% (v/v) formamide with 0.1% bovine serum albumin/0.1% Ficoll/0.1% polyvinylpyrrolidone/50 mM sodium phosphate buffer at pH 6.5 with 750 mM sodium chloride, 75 mM sodium citrate at 42° C.; or (3) employ 50% formamide, 5×SSC (0.75 M NaCl, 0.075 M sodium citrate), 50 mM sodium phosphate (pH 6.8), 0.1% sodium pyrophosphate, 5×Denhardt's solution, sonicated salmon sperm DNA (50 μg/ml), 0.1% SDS, and 10% dextran sulfate at 42° C., with washes at 42° C. in 0.2×SSC (sodium chloride/sodium citrate) and 50% formamide at 55° C., followed by a high-stringency wash consisting of 0.1×SSC containing EDTA at 55° C.
[0154]When a nucleic acid of interest is in "operative association" with a promoter, the promoter is able to direct transcription of the nucleic acid of interest in an appropriate expression system, with the nucleic acid of interest in the correct reading frame for translation. Preferably when a nucleic acid of interest is in operative association with a promoter, the transcript of the nucleic acid of interest contains an appropriately located ribosome binding site for expression in an appropriate expression system of the polypeptide encoded by the nucleic acid of interest. See for example Sambrook et al. (1989) and Ausubel et al. (1992).
[0155]Variants of the genes, coding regions, nucleic acids, polypeptides and promoters specifically disclosed herein preferably have the same function as those specifically disclosed. In relation to the mmyR, mmfR and mmfL genes such function may be encoding an MmyR, MmfR and MmfL polypeptide, respectively; in relation to the MmyR, MmfR and MmfL polypeptides and the repressible promoter (e.g. the mmyTOG promoter, the mmy . . . XCAPK promoter or the mmfLHP promoter) such function may be the ability to interact with each other according to the model proposed above.
[0156]As used herein "mmyTOG" denotes nucleic acid including MmyT, mmyO and mmyG genes. The same applies mutatis mutandis to "mmy . . . XCAPK", the dots indicating the previously unidentified mmyD gene upstream of mmyX in the same transcription unit, and possibly the mmyB, mmyQ and mmyE genes, although these may form a separate transcription unit under the control of a different (mmyBQE) promoter. "mmyBQE" denotes nucleic acid including mmyB, mmyQ and mmyE genes. "mmyBQEDXCAPK" denotes nucleic acid including mmyBQE and mmyDXCAPK. "mmyYF" denotes nucleic acid including mmyY and mmyF genes. "mmf" denotes nucleic acid including mmfP, mmfH, mmfL and mmfR genes. "mmfLHP" denotes nucleic acid including mmfL, mmfH, and mmfP genes. "mmyR-mmf-mmyTOG" denotes nucleic acid including an mmyR gene and mmf and mmyTOG. "mmy . . . XCAPK promoter" may be a promoter which controls the transcription of mmyBQEDXCAPK, or a promoter which controls the transcription of mmyDXCAPK only.
[0157]The invention, in its various aspects, will now be described in detail, with reference to the following figures, in which:
[0158]FIG. 1 shows the origin of the regulatory portion. [0159](a) The genome of Streptomyces coelicolor A3(2) consists of a linear chromosome and two plasmids--the circular SCP2 and the linear SCP1. [0160](b) The methylenomycin production genes form a large gene cluster on the SCP1 plasmid (Chater and Bruton, 1985; Redenbach et al., 1998). [0161](c) About 25 kb of DNA includes regulatory, resistance and biosynthetic genes associated with methylenomycin production. [0162](d) The leftmost ca. 8 kb of the gene cluster comprises regulatory portion of the present invention, i.e. genes involved negatively and positively in regulating levels of methylenomycin production, and promoters under the control of those regulatory genes (it also includes other genes which are transcribed from these promoters). [0163](e) DNA to the right of the regulatory portion shown in (d), comprising the mmr (methylenomycin resistance) gene and genes further to the right, indicating the location of mmyB.
[0164]FIG. 2 shows a comparison of the products of methylenomycin regulatory genes with GBL-binding proteins from various Streptomyces spp. The products of mmyR (mmyrep) and mmfR (mmyrep2) are aligned with GBL-binding proteins associated with the production of virginiamycin (bara, barb), streptomycin (arpa) and showdomycin and mimimycin (fara). Other probable GBL-binding proteins from S. coelicolor A3(2) (cpra, cprb, scbr) and the jadomycin biosynthesis gene cluster (jadr2) are also shown.
[0165]FIG. 3 shows that the product of mmfL (abbreviated here as mmy) is homologous with Streptomyces proteins implicated in the biosynthesis of GBLs. The latter proteins are for biosynthesis of A-factor (afsa), a GBL of S. coelicolor (scba), virginiae butanolide (barx) and IM-2 (farx).
[0166]In FIGS. 2 and 3, dots (".") denote the absence of an amino acid at that position, or the insertion of a gap for optimal sequence alignment and asterisks ("*") denote the end of an amino acid sequence.
[0167]FIG. 4a shows the expression of a foreign gene (xylE) from an expression cassette which comprises the regulatory portion and the foreign gene. [0168]Lower panel: organisation of the methylenomycin gene cluster in a strain engineered to express xylE from the transcription unit containing mmyG. The vector KC861 (Bruton et al, 1991) was engineered to contain the PstI fragment C2.18 (Chater and Bruton, 1983) which is now known to extend from within mmyT to within mmyG: the insert permitted integration of the phage in the configuration indicated. mmyG' and mmy'T denote 3' and 5' truncated copies of those genes, respectively. [0169]Upper panel: time course for catechol oxygenase activity in a strain (based on J1507: Bruton and Chater, 1983) carrying the fusion illustrated. Samples grown on cellophane membranes overland on R2YE were harvested after the indicated culture times, and extracts were made and assayed as in Guthrie and Chater (1990).
[0170]FIG. 4b shows the expression of a foreign gene (xylE) by fusing the foreign gene to different transcription units in the methylenomycin biosynthesis cluster. The fusions were made exactly as in FIG. 4a (lower panel), but with reg or A3.13 fragments (FIG. 9) replacing the C2.18 fragment. [0171]Upper panel: fusion to the mmfLHP transcription unit via the reg fragment; [0172]Lower panel: fusion to the mmy . . . XCAPK transcription unit via fragment A3.13.
[0173]FIG. 5 summarises the results of a Southern blot, demonstrating the extensive deletion of methylenomycin biosynthetic DNA from the R333 mutant. The probe, pIJ518 (Chater and Bruton, 1985) contains a large segment from the centre of the methylenomycin cluster. In a R333 digest (not shown), most of the PstI fragments are missing. Also shown is the organisation of genes within the methylenomycin biosynthetic gene cluster and to the right of the newly sequenced region. Also shown is an EcoRI segment of SCP1 DNA which, when sub-cloned and introduced into an SCP1-S. coelicolor host, stimulated methylenomycin production by an adjacent culture of the indicator "convertor" strain R39 described by Kirby and Hopwood (1977).
[0174]FIG. 6, parts a-d show examples of situations in which the regulatory portion and/or expression cassette could be used to enhance production of useful products. See text for explanations. Preferably in parts b, c and d, either the host strain used contains the mmyB gene, or the mmyB gene is present in the vector containing the expression cassette.
[0175]FIG. 7 shows the entire double-stranded sequence of an approximately 9.5 kb stretch of nucleic acid from the SCP1 plasmid, containing the mmyR, mmfP, mmfH, mmfL, mmfR, mmyT, mmyO, mmyG and mmyJ genes and the start of the mmr (methylenomycin resistance) gene. Some restriction sites are shown. Deduced start and end residues of the genes are as follows:
TABLE-US-00001 Starts at Ends at residue Gene Strand residue number number MmyR bottom 1389 or 1407 796 MmfP bottom 2352 or 2355 1558 MmfH bottom 3554 2352 MmfL bottom 4612 3551 MmfR top 4807 5451 MmyT top 5676 6401 MmyO top 6432 7553 MmyG top 7636 8817 MmyJ bottom 9115 or 9151 8780 Mmr top 9333 not shown
[0176]In the figure:
denotes the start of a gene;[ denotes the end of a gene; and* denotes one of two possible start sites for a gene.
[0177]FIG. 8, parts a-h show the deduced amino acid sequences of the mmyR, mmfP, mmfH, mmfL, mmfR, mmyT, mmyO and mmyG genes, respectively.
[0178]FIG. 9 shows restriction sites flanking DNA fragments used to guide insertion of foreign DNA (such as xylE; FIG. 4) into the mmfHLP, mmyTOG, and mmy . . . XCAPK gene clusters (information from Chater and Bruton, 1993 and 1995, and FIG. 7). Only relevant sites are shown.
[0179]FIG. 10 shows the restriction sites used in a restriction analysis of KC861::C2.18 phages to determine orientation of the C2.18 insert. P=PstI, B=BamHI, SI=SstI, Bg=BglII, r=right hand end, 1=left hand end. Only relevant sites are shown.
[0180]FIG. 11 shows the construction of φG-UP vectors.
[0181]FIG. 12 shows the restriction sites and oligonucleotides used to provide the salIR gene as a BglII fragment suitable for expression in a φG-UP vector.
[0182]FIG. 13 shows in part a the construction of pG-UP. Part b shows the preferred form, pG-UP*, which contains mmyB. The HindIII site of the illustrated version of pG-UP is used to introduce mmyB in a form that leaves a unique HindIII site between the expression cassette and mmyB.
[0183]FIG. 14 shows the use of pG-UP for the expression of the J21 gene set. Numbers on primers refer to base positions in FIG. 7.
[0184]FIG. 15 shows in part a fragment of cosmid cos73, containing the genes from mmyR to mmyG; part b shows this region engineered to terminate with the mmyTOG promoter, for use to control expression of a nucleic acid of interest; and part c shows the construction strategy
EXAMPLE 1
High Levels of Expression of a Foreign Gene Under the Control of Promoters in the Methylenomycin Biosynthetic Gene Cluster
[0185]In order to determine expression levels at different points in the methylenomycin biosynthetic gene cluster (here termed the mmy cluster), derivatives of bacteriophage C31 containing the foreign gene xylE (a gene originating from Pseudomonas: Zukowski et al., 1983) were used to place xylE in defined positions and orientations within the mmy cluster contained in S. coelicolor strains J1507 (which contains the mmy cluster within SCP1NF, a chromosomally integrated copy of SCP1; Bruton and Chater, 1983), or J1506 (a derivative of J1501 (Chater et al. 1982) which has an autonomous copy of SCP1).
[0186]Because KC861, the vector used, lacks the attP site normally used by C31 to permit its integration into the host chromosome during the establishment of lysogeny, it cannot form a prophage by the normal route (Bruton et al., 1991). It is, however possible to provide an alternative integration route by inserting a piece of Streptomyces host DNA into KC861, so that homologous recombination can integrate the prophage at the corresponding position in the host's DNA. Such events, which are quite rare, can be detected if the prophage carries a selectable resistance gene such as vph (viomycin resistance) or tsr (thiostrepton resistance) (Chater & Bruton, 1983; Bruton et al., 1991). In the present case, the mmy inserts placed in KC861 permitted the vector to integrate into particular positions in the mmy DNA of SCP1NF in J1507 or of SCP1 in J1506, with orientations that depended on the orientation of the insert in the vector. pBR327 and pBR322 recombinants (Chater & Bruton, 1983, 1985) were the DNA source for cloning the fragments reg, C2.18, mmr, A4.2 and A3.13 (FIG. 9) into the BamHI site of KC861. The reg fragment was a SstI-BglII subfragment of a larger insertion in pIJ519 (Chater and Bruton, 1985). The other four fragments had PstI boundaries. In order to provide them with BamHI compatible ends, they were introduced into the E. coli plasmid pIJ2925 (Janssen and Bibb, 1993). The mmy inserts were separated from the pBR327/322 vectors by digestion with PstI (or SstI and BglII for reg) and gel electrophoresis and were then ligated to suitably cut pIJ2925. JM101 was transformed with the ligations. The plasmid-host combination allowed blue/white screening for recombinants. For each fragment plasmid DNA of several white colonies was examined by BglII digestion to show whether it contained the insertion. A second enzyme was used to determine its orientation in relation to the polylinker of pIJ2925 (Table 1). This helped later to determine the orientation relative to the xylE gene in the phage vector.
TABLE-US-00002 TABLE 1 The orientation of mmy fragments inserted into pIJ2925 Enzyme used Derivative of to determine Fragment pIJ2925 orientation Orientation* reg pIJ560 XhoI r C2.18 pIJ561 EcoRI r mmr pIJ562 PvuII l A4.2 pIJ563 SstI l A3.13 pIJ564 EcoRI r *"r" indicates that the right end of the insert DNA is located at the right end of the pIJ2925 polylinker; "l" indicates that the left end of the insert DNA is located at the right end of the pIJ2925 polylinker.
[0187]The plasmids pIJ560-pIJ564 were cut with BglII and the purified digested DNA was ligated to KC861 DNA cut with BamHI. Protoplasts of S. lividans 1326 were transfected with the ligated DNA. 100-200 well separated plaques were picked to masterplates of 50 plaques each.
[0188]Phages with the desired insertion of mmy-DNA fragments were identified by a hybridisation signal on plaque lifts. The phage DNA was transferred from the plaques on the masterplate onto nitrocellulose (Benton & Davis, 1977). From the plasmids pIJ560-pIJ564 the inserted DNA fragments were isolated and labelled non-radioactively by the digoxigenin system of Boehringer Mannheim to prepare probe DNA for the hybridization. Four positive plaques on the masterplate for each of the five insertions were purified to get phage suspensions from single plaques. Phage DNA was prepared and analysed by digestion with restriction enzymes to verify that it contained the expected insertion and to determine its orientation. As an example the situation is demonstrated for the insertion of C2.18 in FIG. 10. In this example restriction digests (not shown) of each sample with respectively PstI and SstI were used to determine the orientation of the insert in the sample. Table 2 shows the results of the DNA analysis and gives the names of the constructed phages.
TABLE-US-00003 TABLE 2 Structure of KC861 recombinants with mmy-DNA fused to xylE. Number of Orientation representatives in Inserted Phage of inserted four analysed fragment designation fragment phages reg KC133 r 3 (A, B, C) (2.2 kb) KC134 l 1 C2.18 KC135 r 2 (A, B) (2.05 kb) KC136 l 2 (A, B) mmr KC137 r 3 (A, B, C) (2.5 kb) KC138 l 1 A4.2 KC139 r 1 (2.75 kb) KC140 l 3 (A, B, C) A3.13 KC141 r 3 (A, B, C) (2.28 kb) KC142 l 1 A, B and C name the different isolated phages; r indicates that the right end of the insert DNA is to the right of KC861; and l indicates that the left end of the insert DNA is to the right of KC861.
[0189]The orientation was determined using SstI, for which there is a unique site in the polylinker regions of KC861 and pIJ2925. Information about the orientation of the cloned fragment in pIJ2925 was necessary (see Table 1).
[0190]Phage suspensions from selected single plaques were used to lysogenise the strains J1506 (SCPI+) and J1507 (SCPINF). The integration of the phage conferred thiostrepton resistance on the lysogens and placed the xylE fusions into the mmy gene cluster of the host strain (FIG. 4a; FIG. 6a). To prepare suitable lysogens, 10-20 l of a phage suspension from a single plaque of each of KC133-KC142 was spotted on an R5 plate spread with 107-108 spores of J1507 or J1506. After 5-7 days the cultures had sporulated and were replicated to minimal medium containing 50 gml-1 thiostrepton. After c. four days resistant colonies were streaked on R5 plates containing 5 gml-1 thiostrepton to get single colonies, which were then spread on the same medium to obtain spores of purified lysogens.
[0191]To check that the prophages had integrated at the expected locations, Southern blotting was done. Genomic DNA was prepared from 25 ml YEME cultures of the lysogens containing 4 gml-1 thiostrepton. In each case an XhoI digestion of the DNA was used to investigate disruption in the mmy gene cluster. There is a unique XhoI site in KC861 upstream of the tsr gene. As the XhoI sites flanking or within the cloned fragments were located asymmetrically to their ends, it was possible deduce the orientation of the integrated phage and to confirm the results of the phage analysis. The same non-radioactive DNA probes as for the plaque lifts were used. Table 3 lists the results of the Southern analysis.
TABLE-US-00004 TABLE 3 Southern analysis of lysogens of J1507 and J1506. Distinctive Cloned Distinctive Integrated band for Obtained with fragment band for wt phage disruption J1507 J1506 reg 3.2 kb, 5.2 kb KC133 2.35 kb yes -- KC134 3.7 kb yes -- C2.18 5.25 kb KC135 4.55 kb yes yes KC136 6.7 kb yes yes mmr 6.1 kb KC137 7.75 kb -- yes KC138 4.75 kb yes yes A4.2 6.1 kb KC139 5.25 kb -- yes KC140 7.55 kb yes yes A3.13 2.7 kb KC141 4.4 kb yes yes KC142 4.5 kb -- yes
[0192]Lysogens with the expected hybridisation pattern were obtained with every type of phage, though some of the strains tested did not show the expected pattern. Thus, J1507::KC137(A), 137(B), 139,142(A), 142(B) were thiostrepton resistant, but the integration was not in the right place. As lysogens with the correct construction were obtained for these phages with J1506, no more J1507 lysogens were analysed.
[0193]The BamHI restriction site of KC861 into which the mmy DNA was inserted is part of a multiple cloning site (MCS) located close to, and just upstream of, the promoterless xylE gene. As illustrated by Guthrie and Chater (1990) and Bruton et al. (1991), this has the effect, upon integration of the phage by homologous recombination, of placing xylE under the control of the transcription unit from which the particular insert originated, provided that the transcription unit and xylE have the same orientation. Therefore, the level of expression of the relevant transcription unit is indicated by the level of xylE expression. Expression of xylE is readily monitored because the xylE gene product is an enzyme (catechol 2,3 dioxygenase) that converts colourless catechol into a yellow compound, 2-hydroxymuconic semialdehyde. This can be detected by eye as a yellow zone round colonies after spraying with catechol (the Ylo+ phenotype), or quantitatively by spectrophotometry after cell-free extracts have been prepared (Zukowski et al., 1983; Ingram et al., 1989).
[0194]The in vivo assay for xylE activity of the lysogens (i.e. Ylo+ phenotype) was carried out by spraying catechol onto colonies (Ingram et al., 1989; Bruton et al., 1991). Complete medium (CM; Hopwood et al., 1985), which gave strongest expression, was used throughout the experiments and HMM (Hobbs et al., 1992; solidified with 1% agar), in which the Ylo phenotype was more easily scored but which gave lower expression, was used only occasionally for comparison. Table 4 shows the combined results of repeated xylE tests for all fusion points. Cultures had been sprayed with catechol solution at ages of 42 h and 72 h.
TABLE-US-00005 TABLE 4 Plate assays for xylE activity in J1507 and J1506 lysogens (see following page). Conclusion about Cloned Ylo transcription fragment Lysogen phenotype (see FIG. 9) reg J1507::KC133 -- not rightward across BglII at pos. 13.0 J1507::KC134 +++ leftward across SstI at pos. 10.8 C2.18 J1507::KC135 +++ rightward across PstI at pos. 16.0 J1506::KC134 +++ rightward across PstI at pos. 16.0 J1507::KC136 -- not leftward across PstI at pos. 13.9 J1506::KC136 -- not leftward across PstI at pos. 13.9 mmr J1506::KC137 -- not rightward across PstI at pos. 19.3 J1507::KC138 + leftward across PstI at pos. 16.8 J1506::KC138 + leftward across PstI at pos. 16.8 A4.2 J1506::KC139 -- not rightward across PstI at pos. 22.1 J1507::KC140 ++ leftward across PstI at pos. 19.3 J1506::KC140 -- leftward across PstI at pos. 19.3 A3.13 J1507::KC141 -- not rightward across PstI at pos. 25.0 J1506::KC141 -- not rightward across PstI at pos. 25.0 J1506::KC142 +++ leftward across PstI at pos. 22.7
[0195]Of particular relevance was the finding that the insert in KC135 gave rise to a strong Ylo+++phenotype in strain J1507::KC135. This was examined in closer detail by inoculating ca. 108 J1507::KC135 spores onto each of a series of plates containing CM (Hopwood et al., 1985) supplemented with histidine (50 g ml-1) and uracil (7.5 g ml-1) overlaid with a cellophane disc as described by Tan and Chater (1993), then incubating at 30° C. for the times indicated in FIG. 4a, upper panel, before scraping off the mycelial growth. For catechol 2,3 dioxygenase assays, the mycelium from one cellophane disk was suspended in 0.5 ml extraction buffer, and cell-free extracts were prepared by sonication and clarified by centrifugation as in Ingram et al. (1989). Catechol 2.3 dioxygenase specific activities were determined by spectrophotometry, as above. The results are shown in FIG. 4a, upper panel. The specific activities in samples harvested before 30 h were not significantly above the lowest reliably measurable levels, but between 30 and 40 h, as the growth on the cellophane became dense and morphological differentiation began, there was a very rapid increase in activity up to 30 mU mg-1 protein.
[0196]In an earlier experiment in which xylE was fused to the redX gene, which is involved in the synthesis of another antibiotic in S. coelicolor, Guthrie and Chater (1990) reported peak values of 2 mU mg-1 protein. The mmy-driven transcription of xylE in J1507::KC135 was therefore very strong.
[0197]Further experiments (FIG. 4b) showed a similar pattern and level of xylE expression when suitable fusions were made at other points in the mmy cluster. The positions of these fusions are shown in FIG. 9.
[0198]These results show that a foreign gene (in this example, xylE) can be expressed to quite high levels specifically late in growth when inserted at the illustrated locations in the mmy cluster, without the addition of any inducing agent.
EXAMPLE 2
A Diffusible Substance, Capable of Eliciting Methylenomycin Production from Certain Methylenomycin Non-Producing Mutants, is Produced by a Mutant Containing the Leftmost 6.5 Kb of the Methylenomycin Biosynthetic Gene Cluster, But Little if any Other mmy DNA.
[0199](a) In previous work (Kirby et al., 1975; Kirby and Hopwood, 1977) it was shown that mutants unable to make methylenomycin could be isolated after different procedures. The ability of these mutants to produce and/or respond to extracellular substances relevant to methylenomycin production was tested by growing strains close together on the surface of CM agar (Kirby and Hopwood, 1977). R333 was one of ten mutants that produced an extracellular substance that elicited methylenomycin production by another methylenomycin non-producing mutant, R39. In the work of Kirby and Hopwood (1977), it was considered likely that the substance produced by R333 was converted into methylenomycin. In the present example, DNA was isolated from R333 and digested with the restriction enzymes PstI and PvuII. The digested DNA was subjected to agarose gel electropheresis and blotted onto a nitrocellulose membrane (Southern, 1975). The membrane was then hybridised with a 32P-labelled probe derived by nick translation of pIJ518, a plasmid containing much of the mmy cluster (Chater and Bruton, 1985; FIG. 5). R333 contained a deletion extending rightwards from a position about 6.5 kb inside the left end of the mmy cluster, and ending beyond the righthand end of the KC518 insert. Thus, R333 contains about 6.5 kb of DNA from the left end of the mmy region, and little (perhaps no) other methylenomycin-related DNA (FIG. 5).
[0200]Having discovered the magnitude of the deletion of biosynthetic genes in R333, the present inventors suggest that this 6.5 kb region confers biosynthesis of an extracellular signalling molecule and that this, not an intermediate of methylenomycin biosynthesis, is the substance which is secreted by R333 and which is capable of stimulating R39 to produce methylenomycin.
[0201]This is consistent with the observation that only one of 16 mmy mutants studied by Kirby and Hopwood (1977) could be induced to produce methylenomycin when grown near other mmy mutants.
[0202]The present inventors further suggest that biosynthetic intermediates accumulating in blocked mutants are generally not freely released and exchangeable between strains, but observe that an immediate precursor (desepoxymethylenomycin) is produced by the wild-type and is convertible by SCP1+ strains into methylenomycin (Hornemann and Hopwood, 1978). They further suggest that nearly the whole of the biosynthetic pathway has to be completed before a precursor capable of functioning in cosynthesis is produced--a requirement that would probably necessitate much more than ca. 6.5 kb of biosynthetic genes.
[0203]These suggestions, and other results indicating a regulatory role for some of this DNA (see above), are consistent with the deduced function of genes discovered by sequencing this region of the mmy cluster. The results of this sequence analysis are given in Example 3.
[0204](b) In further confirmation of these predictions, an 8.3 kb EcoRI fragment of SCP1 containing the genes from mmyR to mmyT, with part of mmyO (FIG. 5), was sub-cloned from cosmid 73 of Redinbach et al (1998) into pSET152 (Bierman et al. 1992) and the resulting plasmid was introduced into the φC31 attB site of the SCP1-Streptomyces coelicolor strain J1501 (Kieser et al 2000). The resulting strain, when used in "co-synthesis" tests with R39 (Kirby and Hopwood 1977), elicited methylenomycin production in the R39 strain (as judged by inhibition of the SCP- indicator strain J1501).
EXAMPLE 3
DNA Sequence of the Left End of the Methylenomycin Biosynthetic Gene Cluster
[0205]The DNA sequence of the region from the left end of the mmy region to the previously sequenced (Neal and Chater, 1987) 2.55 kb PstI fragment containing mmr (see FIG. 5) was determined in three sections. The leftmost XhoI fragment (ca. 3.2 kb) was sequenced by dideoxy-sequencing using the method of Sanger et al. (1977), adapted as described in Bruton and Chater (1987). Random sonicated fragments were cloned into M13 mp19 to provide the templates for sequencing, using M13 forward primer (Norrander et al., 1983). The overlapping SstI/PstI (ca. 5 kb) and PvuII (ca. 2.2 kb) fragments were sequenced by automated fluorescence sequencing on an ABI automated sequencer, using templates cloned into pBluescript vectors. For one orientation, templates were generated by the exonuclease III procedure of Henikoff (1984), and in the other, oligonucleotides were designed for "primer walking". All sequences were determined on both strands, and each base position was read through in at least two sequencing reactions. The sequence is given in FIG. 7.
[0206]Use of the FRAME programme (Bibb et al., 1984) led to the recognition that there are nine methylenomycin-related genes to the left of the resistance gene mmr (FIGS. 1 and 5). Only one of these, mmyJ, had previously been sequenced (Neal and Chater, 1987).
Predicted Functions of Genes Identified by Sequencing
[0207]Using the BLAST (Altschul et al., 1990; 1996) and TFASTA (Pearson and Lipmann, 1988) programs to search the major protein and DNA databases, similarities of the deduced products of the eight newly sequenced genes to proteins of known function were discovered. Three of the eight--mmfL, mmfR and mmyR--were of particular interest, because they gave crucial clues about the nature of the regulation of the mmy genes and the putative extracellular signalling molecule, relevant to the design and use of the expression system that is the subject of this patent application. In this section we give details of these similarities. The products of the other five genes all showed some degree of resemblance to various enzymes, and are likely to be directly involved in enzymatic reactions leading to the biosynthesis or metabolism of methylenomycin or the GBL factor involved in regulating methylenomycin biosynthesis.
mmfL
[0208]The predicted product of mmfL (MmfL) showed significant similarity to only four proteins: AfsA, ScbA, BarX and FarX. These proteins are all from other Streptomyces spp., and they are all intimately associated with the production of GBL signalling molecules involved in the regulation of antibiotic production (they are generally believed to be the enzymes responsible for GBL synthesis: Horinouchi et al., 1985, 1989; E. Takano, personal communication). The similarity of MmfL to these proteins is essentially end-to-end, and is very highly significant (FIG. 3). The discovery of such a gene in a small region of DNA that encodes production of an extracellular substance that stimulates methylenomycin production (See Example 2) makes it highly likely that the signal is a GBL, and that MmfL protein encodes a critical step in its biosynthesis.
mmfR and mmyR
[0209]The MmfR and MmyR proteins show significant alignment with a large family of bacterial regulatory proteins (the TetR "superfamily"), principally in a substantial region near the N-terminus of the proteins. This region has been shown in a few of these proteins (Hillen and Berens, 1994) to assume an α-helix-turn-α-helix organisation that permits it to bind to specific sequences in double-stranded DNA.
[0210]The most similar proteins to MmfR and MmyR form a particular sub-group (or "family"). All are from Streptomyces spp., and where sufficiently studied, all are associated with the regulation of antibiotic production and/or morphological differentiation (Onaka and Horinouchi, 1997; Onaka et al., 1997, 1998; Sugiyama et al., 1998; Nakano et al., 1998). The alignments are shown in FIG. 2.
[0211]The greatest similarity is in the region of the DNA-binding domain expected to make sequence-specific contacts with target DNA sequences, leading to the expectation that MmfR and MmyR bind sequences similar to those recognised by the other members of the family (see below). Although the central and C-terminal regions of these proteins are less well conserved, nevertheless there is evidence of similarity throughout these regions, whereas other members of the TetR superfamily do not show similarity to MmfR beyond the N-terminal region. The aligned proteins have a very significant further feature in common--the ones studied to--date all act as specific receptors for different GBL signalling molecules.
[0212]The present inventors have newly discovered that mmfR and the putative GBL biosynthesis gene mmfL are arranged in a similar way, and close inspection of the sequence between them reveals a palindromic sequence (half-site 5'-GGAAGGTATTA-3') that resembles the consensus sequence (half-site 5'-GG(T/C)CGGT(A/T) (T/C)G(T/G)-3') defined for binding of DNA by the ArpA protein that is the archetype of these factor-binding proteins (Onaka and Horinouchi, 1997).
[0213]The present inventors therefore suggest that the chemically undefined extracellular factor secreted by R333 (see Example 2) is a GBL whose extracellular concentration builds up slowly as hyphal density in the cell culture increases, to some critical threshold at which it is effectively perceived by the binding protein encoded by mmfR. This interaction releases MmfR from its location in the bidirectional promoter region, leading to derepression of mmfL and hence of factor biosynthesis. This in turn, it is proposed, causes an acceleration in the rate of factor production, sufficient to interact with, and inactivate, repressor(s) bound either to the mmyTOG and mmy . . . XCAPK promoters or to the promoter of another regulatory gene (e.g. mmyB) needed to activate the mmyTOG and mmy . . . XCAPK promoters, permitting the mmyTOG and mmy . . . XCAPK genes to be expressed. A convenient hypothesis would be that the mmyR gene product (MmyR, see below) is this repressor and that MmyR requires a higher GBL concentration for its repressor function to be inactivated than does MmfR.
EXAMPLE 4
Prevention of Translation of mmfL mRNA is Associated with Failure to Transcribe mmy DNA
[0214](a) In the DNA sequencing in Example 3 mmfL is the only gene to contain a TTA (leucine) codon. Such codons are unexpressed in severe bldA mutants, because bldA encodes the only tRNA capable of translating UUA codons (Leskiw et al., 1991). A prediction of the model for regulation of methylenomycin production is that mmfL mRNA would be untranslatable in a bldA mutant, leading to inability to make the GBL factor and hence to inability to transcribe the genes for methylenomycin production. To test this, xylE fusions into various parts of the mmy cluster of a bldA mutant were constructed using the C31KC861 derivatives described in Example 1. The ability of the resulting strains to express xylE was then tested by the qualitative plate method. Catechol oxygenase activity is not detected, confirming the prediction, and indicating that a TTA-containing gene is involved in regulating mmy gene expression.
[0215](b) In verification of this, the TTA codon of mmfL was changed to CTC in the 0.9 kb SunI internal fragment of mmfL (which was sub-cloned into pFA6a (Wach et al. 1994, replacing the KanMX module), using the "Quick Change" system of Stratagene, followed by reinsertion of the SunI fragment into the 1.7 kb XhoI-BglII fragment containing mmfL (in pIJ2925, Kieser et al. 2000) to give mmfLCTC. This fragment was sub-cloned into pSET151 (Bierman et al. 1992) and introduced into J1703 (bldA-, SCP1NF: Lawlor 1997). Most of the resulting strains acquired the ability to stimulate R39 to produce methylenomycin in co-synthesis, proving that non-translation of the TTA codon of mmfL is responsible for factor non-production in the bldA mutant. However, none of the strains produced methylenomycin, indicating that an additional bldA-dependent step intervenes between factor production and methylenomycin production. This was further verified, since J1703 was not stimulated to produce methylenomyin by growth adjacent to the GBL factor-producing strain R333. The likely intervening step is a gene (mmyB) located about 9 kb to the right of the sequence given in FIG. 7. This gene is predicted to encode a DNA-binding protein and contains a TTA codon.
EXAMPLE 5
Construction of a Vector (φG-UP) Permitting the Easy Insertion of Small Transcription Units into the Expression Region of the Methylenomycin Gene Cluster
[0216]In order to facilitate expression of foreign genes, the vector φG-UP is first constructed. Using the procedures in Hopwood et al. (1985) and Kieser et al. (2000), DNA is extracted from large-scale preparations of φC31 KC889 (FIG. 11). 5 μg of this DNA is digested by EcoRI and--separately--5 μg is digested by XhoI plus SstI. Completeness of digestion is checked by agarose gel electrophoresis (1% agarose) of 0.5 μg of each digested DNA, immediately after heating it to 70° C. for 10 min and cooling it on ice (to separate cohesive ends of the phage DNA).
[0217]After phenol extraction, the two digests are mixed and co-precipitated with ethanol, washed once with 70% ethanol, then dissolved in 100 μl Klenow buffer. The solution is heated at 70° C. for 10 min in a waterbath, which is then turned off and allowed to cool down overnight (this permits φC31 cos ends to join together). The dissolved DNA is then subjected to filling in of the 5' ends generated by XhoI and EcoRI, using Klenow enzyme, before phenol extraction, ethanol precipitation, washing with 70° ethanol, and redissolving in 200 μl ligation buffer, prior to ligation overnight using conditions suitable for blunt end ligation (low ATP, high T4 DNA ligase). After ligation, the DNA is precipitated with ethanol, washed in 70% ethanol, and dissolved in 100 μl TE buffer, prior to being used for transfection of Streptomyces lividans 1326 essentially as described by Hopwood et al. (1985) and Kieser et al. (2000).
[0218]Most plaques are expected to have phages with the desired DNA structure (deletion of the XhoI-EcoRI fragment that contains vph), so screening is done by restriction analysis of DNA isolated from the progeny of 12 of the transfectant plaques. The use of PstI, BglII and/or BamHI provides diagnostic digestion patterns.
[0219]A phage with the correct organisation is identified. A large-scale DNA preparation from this phage is digested with PstI, ethanol precipitated, washed with 70% ethanol and redissolved in TE buffer, before being ligated (at 100-200 μg ml-1 DNA) with an equimolar amount of the 2.05 kb PstI fragment C2.18 (Bruton and Chater, 1983), obtained from pIJ518 by PstI digestion followed by separation by agarose gel electrophoresis and isolation from the gel. After ligation, the DNA is ethanol precipitated, washed and redissolved in 20 μl TE buffer. This solution is used to transfect S. lividans, and the resulting plaques are arrayed on master plates prior to analysis by filter hybridisation (Benton and Davis, 1978) to identify candidates with the desired insertion. The probe for this analysis is the C2.18 PstI fragment, labelled non-radioactively with the digoxigenin system. Twelve candidate plaques are used to propagate phage for small-scale DNA preparation. The DNA is analysed by restriction analysis, using two enzyme combinations: BamHI plus PvuII, and BglII plus PvuII. A phage with each orientation of the insert is retained. Of these, the phage in which the BglII plus PvuII digest gives a 1.2 kb fragment and the BamHI plus PvuII digest gives a 0.8 kb fragment, is termed φG-UP (R) and that in which the BamHI plus PvuII digest gives a 1.2 kb fragment and the BglII plus PvuII digest gives a 0.8 kb fragment is termed φG-UP (L) (FIG. 11).
EXAMPLE 6
Production of the SalI Restriction Enzyme by Placing the salIR Gene Under the Control of the Expression Cassette in a φG-UP Vector
[0220]SalI is a restriction enzyme with substantial use for molecular biology, and therefore with substantial sales. The genes for SalI and the SalI methylase were cloned by Rodicio and Chater (1988) from the producing organism, Streptomyces albus G, and sequenced by Rodicio et al. (1994). They are arranged in tandem, and are expressed as a bicistronic operon (salIR preceding salIM) (Rodicio et al., 1994; Alvarez et al., 1993). In addition, salIM is expressed from its own promoter (Alvarez et al., 1993). Expression of these genes is usually at a very low level, a very high specific activity of SalI being generated by a small amount of protein (of the order of 106 units μg-1 protein). Here we show how the expression cassette in φG-UP can be used to overproduce SalI.
[0221]The salIRM genes are present in pIJ4430 (Rodicio and Chater, 1988). To introduce the gene pair into φG-UP, pIJ34430 is first cleaved with BclI and then with RcaI to generate a fragment with protruding 5' GATC and 5' CATG single-stranded ends. This fragment is inserted into the intermediate vector pIJ2925 (Janssen and Bibb, 1993), using the oligonucleotide adaptors shown in FIG. 12. The fragment is then excised from the intermediate vector by digestion with BglII, and ligated with φG-UP (L) cleaved with BglII. The ligation mixture is used to transfect S. lividans, and the resulting plaques are screened by plaque hybridisation, using the digoxigenin-labelled salIRM BclI-RcaI fragment as a probe. Phages from twelve hybridizing plaques are used for small-scale DNA preparations, and the resulting DNA samples are analysed by digestion with BglII to demonstrate the presence of the full-length 2.9 kb insert, and PstI to determine the orientation of the insert. An additional 2.9 kb PstI fragment would indicate the incorrect orientation, and the absence of such a fragment would indicate the correct orientation.
[0222]Once identified, the desired phage is used to prepare a high titre stock, spots of which are placed on an R2YE plate spread with J1507 spores. After 4-6 days at 30° C., when sporulation has taken place, the plate is replicated to MM (supplemented as necessary for the growth of the auxotrophic J1507) containing thiostrepton (50 μg ml-1), and colonies present after 4 days are purified by single colony isolation then used to prepare confluent plates for the harvesting of dense spore suspensions.
[0223]In order to obtain the desired SalI enzyme, initially on a demonstration scale, the spores are used to inoculate 50 ml CM in a baffled 500 ml flask and the culture is incubated with shaking at 30° C. until stationary phase. At this time, the expression cassette is auto-activated, and the SalI RM genes expressed. The cloned salIM gene will achieve two purposes: the use of its own promoter during early growth will have permitted modification of the host DNA, so rendering it immune to cleavage when SalI is eventually produced; and the expression of SalIM during the main expression period will ensure the optimal production of salIR. Further extraction and purification of SalI follows standard procedures for restriction enzymes.
EXAMPLE 7
(a) Construction of pG-UP, an Integrative Plasmid Vector for the Activation of Cryptic Gene Clusters
[0224]As shown in FIG. 6(b), it is possible to cause the expression cassette to integrate into desired positions in a Streptomyces genome, and thereby to elicit stationary phase expression of adjacent genes. In Example 8, this is put to use in the expression of cryptic genes potentially encoding a new secondary metabolite. Here we describe the construction of pG-UP, an E. coli plasmid containing the expression cassette and capable of transfer into Streptomyces. Insertion of appropriate Streptomyces DNA into pG-UP will permit the use of the construct for gene expression of this kind. The vector is based on pSET151 (Bierman et al., 1992), though any E. coli replicon with a marker permitting selection in Streptomyces and lacking Streptomyces replication or chromosomal integration machinery could be used.
[0225]To construct pG-UP, the 6.2 kb BamHI and BstZ17I fragment of pIJ519 (Chater and Bruton, 1985; and FIG. 7) is isolated from an agarose gel and its ends are blunted by filling in with Klenow enzyme. It is ligated with pSET151 (precleaved with EcoRI and blunted by filling in with Klenow enzyme). After transformation of E. coli strain JM101 transformants are analysed by colony hybridisation using the BamHI and BstZ17I 6.2 kb fragment of pIJ519 as probe to detect the desired insert, and plasmid DNA is extracted from 12 positive colonies. The DNA is digested with BamHI plus BglII to determine the orientation of the insert. An example with a 2.3 kb fragment (rather than a 4 kb fragment) is chosen as pG-UP (FIG. 13a).
[0226]In the form shown in FIG. 13a, the effective use of pG-UP would be guaranteed I na host containing SCP1, to supply the additional genetic component (mmyB) revealed by experiments outlined in Example 4. The use of pG-UP in SCP1- strains will benefit from the further incorporation of mmyB into pG-UP or into the host genome (see Example 7b).
(b) Incorporation of mmyB into pG-UP to Give pG-UP*
[0227]In order to make the effective use of pG-UP independently of a separately provided mmyB gene, mmyB is obtained from the SCP1 plasmid by PCR amplification using the following primers, with cosmid 73 (Redinbach et al 1998) as template:
TABLE-US-00006 5' TATAAGCTTGGTGAACTCCTTCGGCGAGTGGTTCGGA 3' 5' TATGGTACCGGGGAGAACTCCTTGGGATACTTCCTG 3'
[0228]After amplification and conventional preparation for digestion, the PCR product is digested with KpnI, then purified before ligation with the unphosphorylated oligonucleotide 5' AGCTGTAC 31. After gel purification, the linear DNA is cleaved with HindIII, repurified, and ligated with HindIII-cleaved pG-UP. After transformation of E. coli strain DH5 alpha, transformants are screened by colony hybridisation with a mmyB-specific probe, and plasmid is isolated from suitable colonies for verification by restriction analysis for constructs corresponding to FIG. 13b.
EXAMPLE 8
Use of the Expression Cassette in pg-UP in Forced Expression of Genes Apparently Encoding an Unknown Polyketide Molecule
[0229]The project to sequence the genome of Streptomyces coelicolor A3 (2) (www.sanger.ac.uk/Projects/S_coelicolor) has revealed several genes and gene clusters that encode proteins related to some known to be involved in the production of valuable antibiotics. One example is found in cosmid J21, which contains (inter alia) a series of six or seven genes (here termed "the J21 gene set") that appear to form a single transcription unit of perhaps 18 kb, among which two encode probably multidomain β-ketoacyl synthases of the type involved in the biosynthesis of erythromycin, rapamycin, tylosin, avermectin and other macrocyclic polyketides (Hopwood, 1997). No such compound is known to be made by S. coelicolor A3 (2). In order to force expression of the J21 gene set, so that culture fluids can be screened for novel compounds, the J21 gene set is placed under the control of the new expression cassette. For this purpose, a PCR-amplified fragment of J21 DNA is cloned into the pG-UP vector as indicated in FIG. 14. In outline, the fragment is amplified from J21 DNA with the use of an oligonucleotide permitting the start codon of the first gene to be maximally accessible to ribosomes translating mmyG from the expression cassette, and a reverse primer oligonucleotide permitting amplification of a c. 1 kb fragment. Primers include features permitting ready subcloning into G-UP. After transformation of E. coli JM101 and arraying of colonies as patches on a masterplate, colony hybridisation is used to identify colonies containing the J21 insert (with, as probe, the amplified PCR fragment labelled non-radioactively). Plasmid DNA is prepared from candidate colonies by the standard alkaline lysis procedure, and checked by restriction analysis with the enzymes BamHI and HindIII. An example of such a plasmid, in which the orientation of the insert permits its "sense" transcription from the expression cassette, is introduced by transformation into the non-methylating E. coli strain ET12567 (MacNeil et al., 1992) containing the mobilising plasmid, UZ8002 (Flett et al., 1997); and transformants are used in conjugal mating with S. coelicolor M145, selecting thiostrepton-resistant, nalidixic acid-resistant exconjugants (Bierman et al., 1992). Most transformants are expected to contain pG-UP integrated, by homologous recombination, at the start of the J21 gene set. After culture of five representative transformants on R2YE (Hopwood et al., 1985) containing thiostrepton (5 μg/ml), spores are harvested and used to inoculate both liquid and surface CM cultures, which are then incubated for 3 days before extraction with ethyl acetate prior to conventional HPLC and mass spectrometry to determine the structures of any new compounds (e.g. McDaniel et al., 1993).
[0230]In an improved version of this strategy, mmyB is inserted into the polylinker HindIII site of pG-UP, preferably in a form regenerating a HindIII site only between the regulatory cassette and the mmyB gene (see Example 7b and FIG. 13b).
EXAMPLE 9
Fusing Genes to the Cassette in Vectors that can be Maintained in Streptomyces Hosts
[0231]Many antibiotic pathways are highly dependent for expression on pathway-specific transcriptional activators. Additional copies of these genes often stimulate substantial overproduction of the cognate antibiotic (e.g. Chater, 1990). The use of the expression cassette to express such genes would permit over-expression to be confined to dense cultures, thereby minimising antibiotic production during earlier stages of growth: premature production would diminish overall yield and might be lethal if the antibiotic were a novel compound made by a genetically engineered hybrid pathway for which no self-resistance mechanism had evolved. The ability to express such hybrid gene sets in a standard host-vector system would permit the ready screening of large combinational libraries. Examples might include recombinant libraries of type I polyketide synthase genes. In another use of this kind of vector, DNA isolated directly from the environment (e.g. soil) can be expressed from the expression cassette, permitting screening for novel compounds (FIG. 6d). To facilitate this approach, the cassette, prepared as above, can be combined with different vectors capable of stable maintenance in Streptomyces hosts. Examples of such vectors include: those that are maintained as autonomous plasmids, at low copy or medium copy number (usually based on SCP2), or at high copy number (often based on pIJ101); and those that integrate efficiently into the chromosome by site-specific recombination involving the att sites of prophages (such as C31: see FIG. 6d for an example), integrative plasmids (such as pSAM2) or site-specific transposons (such as IS117).
EXAMPLE 10
Providing a Clean Background
[0232]The bldA gene, which can be inactivated without interfering with growth, encodes the tRNA for the rare codon UUA (TTA in the DNA). TTA codons are present in most antibiotic gene clusters, but not in genes for growth. For this reason, bldA mutants make no antibiotics. The expression cassette contains a TTA codon, but a TTA codon-free version has been engineered to permit expression of mmfL in a bldA mutant (see Example 4).
[0233]To allow the effects of this to be full manifested, the TTA codon of mmyB is similarly engineered to an alternative leucine codon, using the Stratagene "Quick Change" system, and the altered gene is introduced by standard procedures into the bldA host strain, along with the expression cassette coupled to the genes to be expressed. In one preferred case, the TTA-free mmyB gene is introduced into a pG-UP vector as indicated in FIG. 13b. However, it can be introduced separately from the expression cassette, e.g. as part of plasmid SCP1. Because nearly all the TTA codons in antibiotic clusters are in regulatory genes, newly discovered sets of genes for biosynthetic enzymes are usually TTA-free. Therefore, expression of such genes from the TTA-free expression cassette will usually be effective in a bldA host. Any new metabolite will be made in the absence of other antibiotics, making it easier to study a range of biological and chemical aspects of the new metabolite without the need to separate it from other bioactive metabolites. Accordingly, the vectors described in examples 5, 7 and 9 would also be constructed with the TTA-free version of the cassette, to permit their use in bldA mutants hosts such as J1703, which contains an integrated copy of SCP1 [for the kind of vector described in example 5], or J1700, which does not contain any DNA from the methylenomycin cluster [for the kinds of vector described in examples 7 and 9].
EXAMPLE 11
Evaluation of the Methylenomycin Promoter
Summary
[0234]The methylenomycin cluster is borne on the large linear plasmid, SCP1, of Streptomyces coelicolor. Previous work has indicated that the promoters within this cluster are strong and could be used commercially.
[0235]This example describes a series of experiments to evaluate the strength of the methylenomycin promoter P.sub.mmyTOG in S. coelicolor and heterologous hosts such as S. lividans and S. erythraea.
[0236]In order to evaluate the promoter, a number of test vectors were engineered in which a reporter gene is placed under the control of the methylenomycin promoter. This fragment was then placed into a suitable vector for introduction into an appropriate host. The reporter gene used was the DEBS1-TE encoding gene. The strength of the promoter can be assessed on the basis of yield of triketide lactone. The actinorhodin promoter P.sub.actI was used as a positive control.
[0237]A gene cassette containing 5 genes, mmyR, mmfP, mmfH, mmfL and mmfR, and the promoter for mmyT, was constructed and is called the promoter cassette. In addition, a separate gene has been identified, mmyB, which contains a rare TTA codon, and is also thought to be involved in the regulation of this promoter. Only trace amounts of triketide lactone are observed when the promoter cassette is used alone in S. coelicolor. Yields increase by 40-100-fold when the plasmid SCP1 is present in the cells. SCP1 harbours the methylenomycin cluster, which contains mmyB. We therefore suggest that the observed increase is due to the presence of the mmyB gene product.
[0238]The experiments carried out in the evaluation of the methylenomycin promoter are described in detail below.
(a) Isolation of the Promoter Cassette
[0239]The final expression vector contains the promoter cassette as a SpeI/NdeI fragment with the NdeI site located such that the ATG start codon of the gene of interest is optimally spaced from the ribosomal binding site of the promoter. This was achieved by amplifying each end of the cassette by PCR using oligonucleotide primers designed to incorporate the sequence for the appropriate restriction enzyme, and cloning in the central region using existing sites (FIG. 1).
[0240]The promoter cassette was isolated from cos73 (Redenbach et al 1998) as follows (see FIG. 15):
[0241]The 8366 bp EcoRI fragment of cos73 was cloned into the unique EcoRI site of pUC18 to give plasmid pCJR332.
[0242]The same EcoRI fragment was used as a template for PCR amplification of the ends of the promoter cassette. oligonucleotide primer CR343 was designed to introduce the SpeI site at 2401-2406 bp (numbering from the beginning of the EcoRI fragment), this is 200 bp after the end of mmyR. Oligonucleotide primer CR344 is fully complementary to the wild-type sequence and binds at a SanDI site (3429-3435 bp--numbering from the beginning of the EcoRI fragment).
TABLE-US-00007 ATTACTAGTTCGCCGAGCGGCTGCGCTCGCTCCGTC CR343 CCGCCGACGCGGGACCCCGCTGTGCAT CR344
[0243]The 1050 bp PCR product was phosphorylated by treatment with T4 polynucleotide kinase and cloned into pUC18 previously digested with SmaI and dephosphorylated. The orientation was determined by restriction enzyme digestion and inserts from a number of clones of the desired orientation were sequenced to check for errors incorporated during in vitro polymerisation. None of the clones were error-free, all errors are substitutions and are within the primer binding region and after the stop codon of mmyR. It was considered that these would not affect the promoter cassette, but as a precaution two of these were selected to carry forward with the experiments and designated pCJR331A and pCJR331B, the errors are described below:
pCJR331A contains a G at 2415 bp where the wild-type sequence has a CpCJR331B contains a G at 2418 bp where the wild-type sequence has a C and a G at 2425 bp where the wild-type sequence has a C
[0244]The 4750 bp SanDI fragment (3431-8180 bp numbering from start of 8366 bp EcoRI fragment) from pCJR332 was cloned into the unique SanDI site in each of pCJR331A and pCJR331B to give plasmids pCJR334A and pCJR334B which were confirmed by restriction analysis.
[0245]A second PCR was used to introduce the NdeI site for cloning of genes under the P.sub.mmyTOG. Oligonucleotide primer CR346 was designed to introduce the NdeI site at 7481-7486 bp (numbering from the beginning of the EcoRI fragment). Oligonucleotide primer CR345 is fully complementary to the wild-type sequence and binds at an XcmI site 6844-6855 bp (numbering) from the beginning of the EcoRI fragment).
TABLE-US-00008 AATCACTGGCCATCGCCGTGGTGGAGGAGCACT CR345 TTTCATATGCGCCCGCGCTCCCAGTCTCTTCTGCCA CR346
[0246]The 657 bp PCR product was phosphorylated by treatment with T4 polynucleotide kinase and cloned into pUC18 previously digested with SmaI and dephosphorylated. The orientation was determined by restriction enzyme digestion and inserts from a number of clones of the desired orientation were sequenced to check for errors incorporated during in vitro polymerisation. One correct clone was selected and designated pCJR328.
[0247]The plasmids pCJR334A and pCJR334B were digested with HindIII and XcmI and the 4491 bp inserts isolated. These were used to ligate into pCJR328 digested with HindIII and XcmI. Correct clones were identified using restriction analysis and designated pCJR335A and pCJR335B.
[0248]These two plasmids, pCJR335A and pCJR335B contain the promoter cassette as defined previously on a SpeI/NdeI fragment. In order to test the utility of this promoter cassette it was introduced into different backbone vectors, which could be used in a number of different hosts, see (b) and (c).
(b) Construction of Plasmids pCMS100 and pCMS101
[0249]The backbone for the first expression vector is pCJR30 (Rowe et al 1998), which has been used previously for the production of the DEBS1-TE triketide lactone, from the actinorhodin promoter P.sub.actI, in Streptomyces coelicolor. The plasmid pCJR30 is therefore the positive control for these experiments and will also provide the backbone for the expression vectors with the methylenomycin promoter.
[0250]pCJR30 was digested with NdeI and SpeI and the promoter cassettes from pCJR335A and pCJR335β isolated as SpeI/NdeI fragments. The promoter cassettes were ligated to the backbones, correct clones identified by restriction analysis, and a single clone from each ligation designated pCMS100A and pCMS100B as appropriate.
[0251]pCMS100A and pCMS100B are final constructs for testing the strength of the methylenomycin promoter P.sub.mmyTOG. These plasmids can be used to assess levels of DEBS1-TE triketide lactone production in actinomycete hosts which can maintain the SCP2* origin of replication.
[0252]The backbone for the second expression vector is pCJR65 (pCJR65 is pCJR24 (Rowe et al 1998) containing DEBS1-TE as an NdeI/XbaI fragment), which has been used previously for the production of the DEBS1-TE triketide lactone, from the actinorhodin promoter P.sub.actI, in Saccharopolyspora erythraea. The plasmid pCJR65 contains no origin of replication for actinomycetes and relies on the presence of the DEBS1-TE encoding gene as homologous DNA to allow integration into the chromosome.
[0253]pCJR65 was digested with NdeI and SpeI and the promoter cassettes from pCJR335A and pCJR335β isolated as SpeI/NdeI fragments. The promoter cassettes were ligated to the backbones, correct clones identified by restriction analysis and a single clone from each ligation designated pCMS101A and pCMS101B as appropriate.
[0254]pCMS101A and pCMS101B are final constructs for testing the strength of the methylenomycin promoter P.sub.mmyTOG. These plasmids are used to assess levels of DEBS1-TE triketide lactone production in S. erythraea JC2 (Rowe et al 1998), or the level of erythromycin production in S. erythraea wild-type.
(c) Construction of Plasmids pCMS104 and pCMS105
[0255]Incorporation of the mmyB gene into pCMS100 and pCMS101 was engineered as follows;
[0256]The mmyB gene was amplified from cos73 with primers CR349 and CR350, which have the following sequences;
TABLE-US-00009 TATAAGCTTGGTGAACTCCTTCGGCGAGTGGTTCGGA CR349 TATAAGCTTGGGGAGAACTCCTTGGGATACTTCCTG CR350
[0257]Each of the oligonucleotide primers has a HindIII site (AAGCTT) incorporated at the 5 prime ends.
[0258]In the published database sequence AJ276673, the mmyB gene is located on the complementary strand between 18032 bp and 18892 bp--the oligonucleotide primers bind in the following positions;
17854-17890 binding region of CR35019095-19122 binding region of CR349
[0259]The total fragment then covers the region 17854-19122 bp with HindIII sites directly flanking this. The entire non-coding DNA from either end of the gene is included in this fragment. It is anticipated that upstream of this gene there will be a promoter and this strategy should ensure that any promoter sequences are incorporated and if any terminator sequences are present these should also be included within the fragment. The PCR fragment was cloned into pUC18 previously digested with SmaI and dephosphorylated, and insert containing clones identified by restriction analysis. The insert was sequenced to confirm that no errors had been incorporated during PCR amplification and the resulting plasmid was called pMMYBH.
[0260]The mmyB gene was then isolated on a HindIII fragment and cloned into HindIII digested, dephosphorylated pCMS100A, pCMS100B, pCMS101A and pCMS101B to give pCMS104A, pCMS104B, pCMS105A and pCMS105B.
(d) Production of the DEBS1-TE Triketide Lactone from the P.sub.mmyTOG Promoter in Streptomyces coelicolor
[0261]The following S. coelicolor strains were transformed with pCMS100A and pCMS100B
S. coelicolor J1501 (SCP1-, SCP2-)S. coelicolor J1506 (SCP1+, SCP2-)S. coelicolor J1508 (SCP1NF, SCP2-)
[0262]Transformants were selected by resistance to thiostrepton and the presence of the plasmid confirmed by re-isolating the plasmid and analysing by restriction digestion.
[0263]Production and analysis experiments were carried out as follows:
[0264]6 ml of YEME+additives (Glycine, MgCl2, uracil and histidine, as recommended)+thiostrepton at a final concentration of 5 μg/litre was inoculated with cells from a plate and grown for 48 hours at 30° C. (in flasks with springs, 250 rpm, 2 inch throw). 300 μl was used as a 5% inoculum into 6 ml of each of YEME and modified Complete Media (as described in Kieser et al 2000 but without the yeast nucleic acid hydrolysate). Cultures were incubated for 88 hours at 30° C. 3 ml of each culture was acidified with formic acid to pH 3 and extracted twice with an equal volume of ethyl acetate, and the organic phase evaporated to dryness using a Buchi rotor evaporator. This crude material was resuspended in 100 μl methanol and 1 μl applied to a Gas Chromatography instrument with a Mass Spectrometry detector. Yields are calculated by comparison to a synthetic standard and the numbers given represent the quantity of triketide lactone in the 3 ml samples removed from the culture.
[0265]Results for production in complete medium:
TABLE-US-00010 Colony S. coelicolor S. coelicolor Plasmid isolate J1501 J1506 pCMS100A 2 No product 3 Trace amount pCMS100B 4 Trace amount pCMS100A 6 17 mg/litre 7 2.3 mg/litre pCMS100B 8 0.5 mg/litre
[0266]These results indicate that there is considerable colony to colony variability in production level, this is to be expected following protoplast transformation. In the absence of SCP1, very little product is observed, a trace amount indicates less than 0.1 mg/litre. We are confident that the trace amounts we see are the triketide lactone as the behaviour of the triketide lactone on GC-MS is predictable and the mass-spectroscopy fragmentation is characteristic. The production levels when cultures were grown in complete medium are comparable to when the same colonies are grown in YEME (full data not yet available). For example, S. coelicolor J1506-[pCMS100A] colonies 6 and 7 yielded 20 and 4 mg/litre respectively when cultured in YEME.
[0267]Further experiments were carried out as follows. Cultures were grown in 6 ml of an appropriate medium and this was used as a 5% inoculum for production cultures. In this way the production cultures for a single isolate should be comparable, and comparison of production levels between isolates should be relatively robust. In all cases yields are based on 3 or 4 ml of culture withdrawn from the flasks, this does not in any of the cases take into account evaporative loss of liquid during fermentation. This means that the yields quoted are yields in the final cultures rather than production levels. Yields are calculated using mass spectrometry, which has an associated error, but such errors should be similar for all samples, with the largest errors being in calculation of the lowest yields due to ill-defined small peaks.
[0268]It should also be noted that the results are always given for the propionate starter triketide lactone [1]. There is a second product from the DEBS1-TE system and this is the equivalent acetate starter triketide lactone [2] which is mainly seen in high producers, or when propionate is limiting in the system. In general, in these experiments this represents less than 5% of the overall product. However, from plates and from some of the complex media there is a significant percentage of this product and in these cases a yield has been calculated based on the propionate starter standard.
(e) Production of the DEBS1-TE Triketide Lactone from the P.sub.mmyTOG Promoter in Streptomyces coelicolor in Different Media
TABLE-US-00011 Culture Yeme Complete Hobbs SSDM 1 J1501[pCJR30]_12 No trace 0.7 no (0.7) 2 J1501[pCMS100A]_11 1 trace 1.1 no 3 J1501[pCMS100A]_12 trace trace 1.4 no 4 J1501[pCMS100B]_7 trace trace no no 5 J1506[pCJR30]_10 No trace trace no (0.07) 6 J1506[pCMS100A]_3 20 17 0.02 0.5 7 J1506[pCMS100A]_9 4 2.3 0.03 0.6 8 J1506[pCMS100B]_7 trace 0.5 trace no Yeme = Yeme + appropriate additives: glycine, MgCl2, uracil and histidine, as recommended. Complete = As described in Kieser et al (2000) but without the yeast nucleic acid hydrolysate. Hobbs = As described in Hobbs et al (1992) SSDM = As described in Caffery et al (1992)
[0269]In all cases yield of triketide lactone are given as 5 mg/litre on the basis of comparison to a known quantity of a synthetic by mass spectrometry. Quantitation by mass spectrometry is subject to a certain amount of error in that the concentration of a molecule in a mixture and the composition of the mixture will effect the ionisation of the subject compound. No indicates that no product was observed by mass spec, and trace indicates that a small peak is observed, but it is not well enough defined to accurately integrate.
[0270]The variability of production levels from one medium to another is significant; and it is proposed that maximal production levels may not have been attained. This can be achieved by statistical analysis production levels as different components of the media are used, and at different concentrations.
[0271]In Hobb's medium, which is adapted for methylenomycin production (i.e. 10 g/litre glucose is added instead of 2), there appears to be higher production in the absence of SCP1.
[0272]The increase in production with SCP1 present appears to be a real effect, despite the small sample size. It is suggested that by providing the mmyB gene in isolation the absolute yield may increase.
(f) Production of the DEBS1-TE Triketide Lactone from the P.sub.mmyTOG Promoter in Saccharopolyspora erythraea in Different Media
[0273]Saccharopolyspora erythraea NRRL2338 JC2 (Rowe et al 1998) was transformed with pCMS100A, pCMS100B, pCMS105A and pCMS105B. Transformants were selected using thiostrepton (final concentration 50 μg/litre) and a secondary round of selection, involving isolating spores and filtering onto fresh selection plates was performed to insure that the isolates contained the resistance from the plasmid.
[0274]Three isolates from each of S. erythraea JC2/pCMS100A and S. erythraea JC2/pCMS100B were used to inoculate 6 ml of TSB+tsr (final concentration 5 μg/litre). These precultures were used to inoculate 6 ml of each of two different production media which have previously been shown to yield good levels of erythromycin, SSDM is a defined minimal medium, and SM3 (Ranganathan et al 1999) is more complex. Production cultures were grown for 7 days and 4 ml of each taken and extracted. Analysis by GC-MS gave the following results.
TABLE-US-00012 SSDM SM3 Culture [1] [1] [2] S. ery 0.5 8.4 3.0 JC2[pCMS100A]_1 S. ery 0.3 8.2 0.2 JC2[pCMS100A]_2 S. ery 0.6 4.0 0.2 JC2[pCMS100A]_3 S. ery 0.8 7.3 0.2 JC2[pCMS100B]_1 S. ery 0.9 5.5 0.2 JC2[pCMS100B]_2 S. ery 0.5 5.0 0.2 JC2[pCMS100B]_3 [1] and [2] refer to the two triketide starter lactones, as described above.
[0275]This experiment demonstrates that the expression cassette can be used to drive expression of a nucleic acid of interest in host cells other than S. coelicolor. Some expression has also been demonstrated in S. lividans.
[0276]Again it is observed that the composition of the media has a significant effect on production level and it is expected that higher yields may be obtained upon optimisation and/or in the presence of mmyB.
(g) a Quick Look at Comparative Yields of Triketide Lactone from Patches on Plates
[0277]To try to get a quick indication of whether or not the mmyB gene would affect production levels, plugs were taken from patches on R2YE plates and extracted to look for triketide lactone product.
[0278]Using strain J1501, which lacks the SCP1 plasmid (and hence a native mmyB gene), a dramatic (up to orders of magnitude) increase in yield was shown between pCMS100A (which lacks mmyB) and pCMS104 (the equivalent plasmid also possessing mmyB).
[0279]Using strain J1506 (which possesses native SCP1 and mmyB), more expression was shown with pCMS100A than was shown using this plasmid in J1501, but less expression was shown with pCMS104 in J1506 than in J1501. Expression in J1506 was similar with both plasmids.
[0280]This is consistent with the mmyB gene product being advantageous for expression, but also with such product being sequestered by (e.g. promoters of) the native SCP1 plasmid when present, rather than acting to increase expression from the expression cassette.
[0281]Yields represent the total product obtained from the whole agar plug (μg).
TABLE-US-00013 Yield (μg) R2YE Culture [1] [2] 26 J1501[pCMS100A]_6 trace none 27 J1506[pCMS100A]_8 1.0 0.4 31 J1501[pCMS104]_2 12 20 32 J1501[pCMS104]_6 1.5 2.5 33 J1506[pCMS104]_1 2.3 2.6 34 J1506[pCMS104]_7 1 1.0
[0282]In some cases the second product was the predominant product, probably reflecting substrate availability, which may therefore be a limiting factor.
REFERENCES
[0283]Aguilar, A. and D. A. Hopwood. 1982, Determination of methylenomycin A synthesis by the pSV1 plasmid from Streptomyces violaceus-ruber SANK95570. J. Gen. Microbiol. 128:1893-1901. [0284]Altschul, S. F., W. Gish, W. Miller, E. W. Meyers and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-410. [0285]Altschul et al. 1996. Methods in Enzymology, 266:460-480. [0286]Alvarez, M. A., K. F. Chater and M. R. Rodicio. 1993. Complex transcription of an operon encoding the SalI restriction-modification system of Streptomyces albus G. Mol. Microbiol. 8:243-252. [0287]Ausubel, F. et al. 1992. Short Protocols in Molecular Biology, Second Edition. Eds, John Wiley & Sons. [0288]Benton, W. D. and R. W. Davis. 1977. Screening lambda gt recombinant clones by hybridization to single plaques in situ. Science 196:180-182. [0289]Bibb, M. J., P. R. Findlay and M. W. Johnson. 1984. The relationship between base composition and codon usage in bacterial genes and its use for the simple and reliable identification of protein-coding sequences. Gene 30:157-166. [0290]Bibb, M. J., J. L. Schottel and S. N. Cohen. 1980. A DNA cloning system for interspecies gene transfer in antibiotic-producing Streptomyces. Nature 284:526-531. [0291]Bierman, M., R. Logan, K. O'Brien, E. T. Seno, R. N. Rao and B. E. Schoner. 1992. Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene 116:43-49. [0292]Bruton, C. J., E. P. Guthrie and K. F. Chater. 1991. Phage vectors that allow monitoring of secondary metabolism genes in Streptomyces. Bio/Technology 9:652-656. [0293]Bruton, C. J. and K. F. Chater. 1987. Nucleotide sequence of IS110, an insertion sequence of Streptomyces coelicolor A3(2). Nucl. Acids Res. 15:7053-7065. [0294]Caffery, P., Bevitt, D. J., Staunton, J., Leadlay, P, F. 1992. Identification of DEBS1, DEBS2 and DEBS3, the multienzyme polypeptides of the erythromycin-producing polyketide synthase from Saccharopolyspora erythraea. FEBS Let. 304, 225-228. [0295]Chater, K. F. C. J. Bruton, A. A. King and J. E. Saurez. 1982. The expression of Streptomyces and Escherichia coli drug resistance determinants cloned into the Streptomyces phage C31. Gene 19:21-23. [0296]Chater, K. F. and C. J. Bruton. 1983. Mutational cloning in Streptomyces and the isolation of antibiotic production genes. Gene 26:67-78. [0297]Chater, K. F. and C. J. Bruton. 1985. Resistance, regulatory and production genes for the antibiotic methylenomycin are clustered. EMBO J. 4:1893-1897. [0298]Chater, K. F. 1990. The improving prospects for yield increase by genetic engineering in antibiotic-producing streptomycetes. Bio/Technology 8:115-121. [0299]Fernandez-Moreno, M. A. 1991. The act cluster contains regulatory and antibiotic export genes, direct targets for control by the bldA tRNA gene of Streptomyces. Cell 66:769-780. [0300]Fisher, S. E., C. J. Bruton and K. F. Chater. 1987. The glucose kinase gene of Streptomyces coelicolor and its use in selecting deletions from defined end-points. Mol. Gen. Genet. 206:35-44. [0301]Flett, F., Mersinias, V. and C. P. Smith (1997) High efficiency intergeneric transfer of plasmid DNA from Escherichia coli to methyl DNA-restricting Streptomyces. FEMS Microbiol. Lett. 155:223-229. [0302]Guthrie, E. P. and K. F. Chater. 1990. The level of a transcript required for production of a Streptomyces coelicolor antibiotic is conditionally dependent on a tRNA gene. J. Bacteriol. 172:6189-6193. [0303]Henikoff, S. 1984. Unidirectional digestion with exonuclease III creates targeted breakpoints for DNA sequencing. Gene 28:351-359. [0304]Hillen, W. and C. Berens. 1994. Mechanisms underlying expression of Tn10 encoded tetracycline resistance. Annu. Rev. Microbiol. 48:345-369. [0305]Hobbs, G. A. I. C. Obanye, J. Petty, J. C. Mason, E. Barratt, D. C. J. Gardner, F. Flett, C. P. Smith, P. Broda and S. G. Oliver. 1992. An integrated approach to studying regulation of production of the antibiotic methylenomycin by Streptomyces coelicolor A3 (2). J. Bacteriol. 174:1487-1494. [0306]Hopwood, D. A. 1997. Genetic contributions to understanding polyketide synthases. Chem. Rev. 97:2465-2497. [0307]Hopwood, D. A., M. J. Bibb, K. F. Chater, T. Kieser, C. J. Bruton, H. M. Kieser, D. J. Lydiate, C. P. Smith, J. M. Ward and H. Schrempf. 1985. Genetic manipulation of Streptomyces. A laboratory manual (Norwich: John Innes Foundation). [0308]Horinouchi, S., H. Suzuki, M. Nishiyama and T. Beppu. 1989. Nucleotide sequence and transcriptional analysis of the Streptomyces griseus gene (afsA) responsible for A-factor biosynthesis. J. Bacteriol. 171:1206-1210. [0309]Horinouchi, S., Y. Kumada and T. Beppu. 1984. Unstable genetic determinant of A-factor biosynthesis in streptomycin-producing organisms: cloning and characterization. J. Bacteriol. 158:481-487. [0310]Hornemann, U. and D. A. Hopwood. 1978. Isolation and characterization of desepoxy-4-5-didehydro-methylenomycin A, a precursor of the antibiotic methylenomycin A in SCP1+ strains of Streptomyces coelicolor A3(2). Tetrahed. Lett. 33:2977-2978. [0311]Ingram, C., M. Brawner, P. Youngman and J. Westpheling. 1989. xylE functions as an efficient reporter gene in Streptomyces spp: use for the study of galP1, a catabolite-controlled promoter. J. Bacteriol. 171:6617-6624. [0312]Janssen, G. R. and M. J. Bibb. 1993. Derivatives of pUC18 that have BglII sites flanking a modified multiple cloning site and that retain the ability to identify recombinant clones by visual screening of Escherichia coli colonies. Gene 124:133-134. [0313]Kieser, T. et al. 2000. Practical Streptomyces genetics. John Innes Foundation. Norwich, UK. [0314]Kirby, R. and D. A. Hopwood. 1977. Genetic determination of methylenomycin synthesis by the SCP1 plasmid of Streptomyces coelicolor A3 (2). J. Gen. Microbiol. 98:239-252. [0315]Kirby, R., L. F. Wright and D. A. Hopwood. 1975. Plasmid-determined antibiotic synthesis and resistance in Streptomyces coelicolor. Nature 254:265-267. [0316]Lawlor, E. J. 1987. Molecular genetics of BldA, a developmental gene of Streptomyces coelicolor. PhD thesis. University of East Anglia. [0317]Leskiw, B. K., E. J. Lawlor, J. M. Fernandez-Abalos and K. F. Chater. 1991. TTA codons in some genes prevent their expression in a class of developmental, antibiotic-negative Streptomyces mutants. Proc. Natl. Acad. Sci., USA 88:2461-2465. [0318]MacNeil, D. J. K. M. Gewain, C. L. Ruby, G. Dazeny, P. H. Gibbons and T. MacNeil. 1992. Analysis of Streptomyces avermitilis genes required for avermectin biosynthesis utilizing a novel integration vector. Gene 111:61-68. [0319]McDaniel, R., S. Ebert-Khosla, D. A. Hopwood and C. Khosla. 1993. J. Am. Chem. Soc. 115:11671-11675. [0320]Nakano, H., E. Takehara, T. Nihira and Y. Yamada. 1998. Gene replacement analysis of the Streptomyces virginiae barA gene encoding the butyrolactone autoregulator receptor reveals that BarA acts as a repressor in virginiamycin biosynthesis. J. Bacteriol. 180:3317-3322. [0321]Neal, R. J. and K. F. Chater. 1987. Nucleotide sequence analysis reveals similarities between proteins determining methylenomycin A resistance in Streptomyces and tetracycline resistance in eubacteria. Gene-58:229-241. [0322]Norrander, J., T. Kempe and J. Messing. 1983. Construction of improved M13 vector using oligonucleotide-directed mutagenesis. Gene 26:101-106. [0323]Onaka, H. and S. Horinouchi. 1997. DNA-binding activity of the A-factor receptor protein and its recognition DNA sequences. Mol. Microbiol. 24:991-1000. [0324]Onaka, H., M. Sugiyama and S. Horinouchi. 1997. A mutation at proline-115 in the a-factor receptor protein of Streptomyces griseus abolishes DNA-binding ability but not ligand-binding ability. J. Bacteriol. 179:2748-2752. [0325]Onaka, H., T. Nakagawa and S. Horinouchi. 1998. Involvement of two A-factor receptor homologues in Streptomyces coelicolor A3(2) in the regulation of secondary metabolism and morphogenesis. Mol. Microbiol. 28:743-753. [0326]Onaka, H., N. Ando, T. Nihira, Y. Yamada, T. Beppu and S. Horinouchi. 1995. Cloning and characterization of the A-factor receptor gene from Streptomyces griseus. J. Bacteriol. 177:6083-6092. [0327]Pearson, W. R. and D. J. Lipman. 1988. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. USA 85:2444-2448. [0328]Ranganathan, A., Timoney, M., Bycroft, M., Cortes, J., Thomas, I. P., Wilkinson, B., Kellenberger, L., Hanefeld, U., Galloway, I. S., Staunton, J. and Leadlay, P. F. 1999. Knowledge-based design of bimodular and trimodular polyketide synthases based on domain and module swaps: A route to simple statin analogues. Chem&Biol. 6, 731-741. [0329]Redenbach, M., K. Ikeda, M. Yamasaki and H. Kinashi. 1998. Cloning and physical mapping of the EcoRI fragments of the giant linear plasmid SCP1. J. Bacteriol. 180:2796-2799. [0330]Rodicio, M. R., T. Quinton-Jager, L. S. Moran, B. E. Slatko and G. G. Wilson. 1994. Organization and sequence of the SalI restriction-modification system. Gene 151:167-172. [0331]Rowe, C. J., Cortes, J., Gaisser, S., Staunton, J., Leadlay, P. F. (1998) Construction of new vectors for high-level expression in actinomycetes. Gene, 216, 215-223. [0332]Rodicio, M. R. and K. F. Chater. 1988. Cloning and expression of the SalI restriction-modification genes of Streptomyces albus G. Mol. Gen. Genet. 213:346-353. [0333]Sanger, F., S, Nicklen and A. R. Coulson. 1977. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 74:5463-5467. [0334]Southern, E. M. 1975. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J. Mol. Biol. 98:503-517. [0335]Sugiyama, M., H. Onaka, T. Nakagawa and S. Horinouchi. 1998. Site-directed mutagenesis of the A-factor receptor protein: Val-41 important for DNA-binding and Trp-119 important for ligand-binding. Gene 222:133-144. [0336]Tan, H. and K. F. Chater. 1993. Two developmentally controlled promoters of Streptomyces coelicolor A3(2) that resemble the major class of motility-related promoters in other bacteria. J. Bacteriol. 175:933-940. [0337]Wach, A. et al. 1984. New heterologous modules for classical or PCR based gene disruptions in Saccharomyces cerevisiae. Yeast 10:1793-1808. [0338]White, J. and Bibb, M. 1997. BldA dependence of undecylprodigiosin production in Streptomyces coelicolor A3(2) involves a pathway-specific regulatory cascade. J. Bacteriol. 179:627-633 [0339]Zukowski, M. M., D. F. Gaffney, D. Speck, M. Kauffmann, A. Findeli, A. Wisecup and J. P. Lecocq. 1983. Chromogenic identification of genetic regulatory signals in Bacillus subtilis based on expression of a cloned Pseudomonas gene.
Proc. Natl. Acad. Sci. USA 80:1101-1105.
Sequence CWU
1
35111DNAArtificialConsensus sequence 1ggycggtwyg k
11211DNAStreptomyces coelicolor
2ggaaggtatt a
113216PRTStreptomyces sp. 3Leu Thr Pro Lys Gln Glu Arg Ala Phe Arg Thr
Arg Thr Gln Leu Val1 5 10
15Leu Ser Ala Ala Glu Ala Phe Asp Arg Gln Gly Phe Ala Thr Ala Ser
20 25 30Leu Thr Ala Ile Ser Asn Ser
Ala Gly Val Ser Asn Gly Ala Leu His 35 40
45Phe His Phe Glu Ser Lys Glu Ala Leu Ala Ala Ala Val Glu Ala
Glu 50 55 60Ala Ala Glu Arg Met Arg
Thr Ile Val Asp Gly Ala Ala Arg Arg Gly65 70
75 80Ala Ser Ala Leu Gln Ala Leu Val Asp Thr Ser
His Ala Val Met Leu 85 90
95Arg Leu Arg Gln Asp Val Val Val Arg Ala Gly Phe Arg Leu Ser Gly
100 105 110Asp Ala Ala Arg Gln Ala
Thr His Asp Leu Pro Glu His Trp Arg Gln 115 120
125Ser Val Val Arg Leu Leu Glu Arg Ala Gly Arg Asp Gly Ser
Leu Thr 130 135 140Ser Ala Val Thr Pro
Ser Asp Val Ala Gly Val Val Thr Ala Thr Val145 150
155 160Leu Gly Phe Gly Val Leu Ala Arg Phe Asp
Ser Ala Trp Leu Ala Ser 165 170
175Gly Ser Leu Ser Gly Phe Trp Lys Leu Met Leu Pro Met Ile Ala Ala
180 185 190Gly Pro Val Glu Arg
Gly Glu Leu Asp Cys Arg Pro Ala Val Pro Ala 195
200 205Asp Val Arg Arg Ala Pro Ala Val 210
2154196PRTStreptomyces sp. 4Met Thr Lys Gln Glu Arg Ala Thr Arg Thr
Arg Asp Ala Leu Ile Lys1 5 10
15Ser Ala Ala Arg Glu Phe Asp Glu His Gly Tyr Ala Leu Ala Lys Leu
20 25 30Ser Ala Ile Ser Ser Gly
Ala Gly Val Ser Pro Gly Ala Leu His Phe 35 40
45His Phe Glu Asn Lys Val Ala Ala Ala Val Glu Ile Asp Ala
Ser Thr 50 55 60Thr Leu Arg Arg Thr
Ala Arg Ile Val Tyr His Gln Arg Ser Asn Ala65 70
75 80Leu Gln Asn Leu Ala Asp Thr Thr His Ala
Leu Ala Arg Leu Val Arg 85 90
95Glu Asp Val Val Val Arg Ala Gly Phe Arg Leu Ser Cys Ser Gln Leu
100 105 110Cys Gly Thr Asp Leu
Asn Leu Arg Gln Glu Trp Gln Ser Cys Val Gln 115
120 125Gln Arg Leu Ala Glu Ala Ala Asp Glu Gly Leu Leu
Ala Ser Asp Ile 130 135 140Gly Gly Gln
Gln Asp Leu Ala Arg Thr Ile Val Ala Ala Thr Ile Gly145
150 155 160Leu Glu Ala Leu Cys Arg Asp
Asn Gly Glu Trp Leu Ser Pro Gly Thr165 170
175Val Thr Gly Leu Trp Arg Thr Leu Leu Pro Ile Val Ala Ala Pro Gly180
185 190Arg Ser Pro Pro1955215PRTStreptomyces
sp. 5Met Ala Arg Gln Leu Arg Ala Glu Arg Thr Arg Ala Thr Ile Val Arg1
5 10 15Ala Ala Ala Asp Leu
Phe Asp Arg His Gly Tyr Glu Ser Thr Ser Leu 20
25 30Ser Glu Ile Val Ala His Ala Gly Val Thr Lys Gly
Ala Leu Tyr Phe 35 40 45His Phe
Ala Ala Lys Glu Asp Leu Ala His Ala Ile Leu Glu Ile Gln 50
55 60Ser Arg Thr Ser Arg Arg Leu Ala Lys Asp Leu
Asp Gly Arg Gly Tyr65 70 75
80Ser Ser Leu Glu Ala Leu Met Arg Leu Thr Phe Gly Met Ala Arg Leu
85 90 95Cys Val Gln Gly Pro
Val Leu Arg Ala Gly Thr Arg Leu Ala Thr Ala 100
105 110Gly Val Pro Val Arg Pro Pro Leu Pro His Pro Phe
Thr Asp Trp Arg 115 120 125Glu Ile
Ala Thr Ser Arg Leu Leu Asp Ala Val Arg Gln Ser Asp Val 130
135 140His Gln Asp Ile Asp Val Asp Ser Val Ala His
Thr Leu Val Ser Ser145 150 155
160Val Val Gly Thr Cys Val Val Gly Gly Thr Leu Glu Pro Ala Gly Arg
165 170 175Gln Pro Arg Arg
Leu Ala Glu Met Trp Tyr Ile Leu Ile Arg Gly Met 180
185 190Val Pro Val Thr Arg Arg Ala Arg Tyr Val Thr
Leu Ala Ala Arg Leu 195 200 205Glu
Gln Glu Thr Gly Thr Thr 210 2156215PRTStreptomyces sp.
6Met Ala Arg Gln Leu Arg Ala Glu Gln Thr Arg Ala Thr Ile Ile Gly1
5 10 15Ala Ala Ala Asp Leu Phe
Asp Arg Arg Gly Tyr Glu Ser Thr Thr Leu 20 25
30Ser Glu Ile Val Ala His Ala Gly Val Thr Lys Gly Ala
Leu Tyr Phe 35 40 45His Phe Ala
Ala Lys Glu Asp Leu Ala His Ala Ile Leu Glu Ile Gln 50
55 60Ser Arg Thr Ser Arg Arg Leu Ala Lys Asp Leu Asp
Gly Arg Gly Tyr65 70 75
80Ser Ser Leu Glu Ala Leu Met Arg Leu Thr Phe Gly Met Ala Arg Leu
85 90 95Cys Val Gln Gly Pro Val
Leu Arg Ala Gly Leu Arg Leu Ala Thr Ala 100
105 110Gly Val Pro Val Arg Pro Pro Leu Pro His Pro Phe
Thr Glu Trp Arg 115 120 125Glu Ile
Ala Thr Ser Arg Leu Leu Asp Ala Val Arg Gln Ser Asp Val 130
135 140His Gln Asp Ile Asp Val Asp Ser Val Ala His
Thr Leu Val Cys Ser145 150 155
160Val Val Gly Thr Arg Val Val Gly Gly Thr Leu Glu Pro Ala Gly Arg
165 170 175Glu Pro Arg Arg
Leu Ala Glu Met Trp Tyr Ile Leu Ile Arg Gly Met 180
185 190Val Pro Val Thr Arg Arg Ala Arg Tyr Val Thr
Leu Ala Ala Arg Leu 195 200 205Glu
Gln Glu Thr Gly Thr Ala 210 2157221PRTStreptomyces sp.
7Val Ala Glu Gln Val Arg Ala Ile Arg Thr Arg Gln Ala Ile Leu Ser1
5 10 15Ala Ala Ala Arg Val Phe
Asp Glu Arg Gly Tyr Gln Ala Ala Thr Ile 20 25
30Ser Glu Ile Leu Thr Val Ala Gly Val Thr Lys Gly Ala
Leu Tyr Phe 35 40 45His Phe Gln
Ser Lys Glu Asp Leu Ala Gln Gly Val Leu Thr Ala Gln 50
55 60Asn Glu Asp Leu Leu Leu Pro Glu Arg Pro Ala Lys
Leu Gln Glu Val65 70 75
80Val Asp Ala Val Met Leu His Thr His Arg Leu Arg Thr Asn Pro Met
85 90 95Val Arg Ala Gly Val Arg
Leu Ser Leu Asp Val Asn Ala Gly Gly Leu 100
105 110Asp Arg Ser Ala Pro Phe Arg Asn Trp Val Asp Lys
Phe Thr Asp Leu 115 120 125Leu Glu
Lys Ala Gln Ala Gln Gly Glu Leu Leu Pro His Val Val Pro 130
135 140Ala Glu Thr Ala Asp Val Ile Thr Gly Ala Tyr
Gly Gly Val Gln Ser145 150 155
160Met Ser Gln Ala Leu Thr Glu His Gln Asp Leu Gly Gln Arg Val Asn
165 170 175Ala Leu Leu Arg
His Leu Met Pro Ser Ile Ala Gln Pro Ser Val Leu 180
185 190Ala Ser Leu His Leu Gly Glu Ser Arg Ala Glu
Glu Val Tyr Leu Glu 195 200 205Ala
Arg Gln Leu Ala Arg Glu Gln Ala Asp Glu Glu Asp 210
215 2208215PRTStreptomyces sp. 8Met Ala Lys Gln Asp Arg
Ala Ile Arg Thr Arg Gln Thr Ile Leu Asp1 5
10 15Ala Ala Ala Gln Val Phe Glu Lys Gln Gly Tyr Gln
Ala Ala Thr Ile 20 25 30Thr
Glu Ile Leu Lys Val Ala Gly Val Thr Lys Gly Ala Leu Tyr Phe 35
40 45His Phe Gln Ser Lys Glu Glu Leu Ala
Leu Gly Val Phe Asp Ala Gln 50 55
60Glu Pro Pro Gln Ala Val Pro Glu Gln Pro Leu Arg Leu Gln Glu Leu65
70 75 80Ile Asp Met Gly Met
Leu Phe Cys His Arg Leu Arg Thr Asn Val Val 85
90 95Ala Arg Ala Gly Val Arg Leu Ser Met Asp Gln
Gln Ala His Gly Leu 100 105
110Asp Arg Arg Gly Pro Phe Arg Arg Trp His Glu Thr Leu Leu Phe Leu
115 120 125Leu Asn Gln Ala Lys Glu Asn
Gly Glu Leu Leu Pro His Val Val Thr 130 135
140Thr Asp Ser Ala Asp Leu Tyr Val Gly Thr Phe Ala Gly Ile Gln
Val145 150 155 160Val Ser
Gln Thr Val Ser Asp Tyr Gln Asp Leu Glu His Arg Tyr Ala
165 170 175Leu Leu Gln Lys His Ile Leu
Pro Ala Ile Ala Val Pro Ser Val Leu 180 185
190Ala Ala Leu Asp Leu Ser Glu Glu Arg Gly Ala Arg Leu Ala
Ala Glu 195 200 205Leu Ala Pro Thr
Gly Lys Asp 210 2159232PRTStreptomyces sp. 9Met Ala
Val Arg His Glu Arg Val Ala Val Arg Gln Glu Arg Ala Val1 5
10 15Arg Thr Arg Gln Ala Ile Val Arg
Ala Ala Ala Ser Val Phe Asp Glu 20 25
30Tyr Gly Phe Glu Ala Ala Thr Val Ala Glu Ile Leu Ser Arg Ala
Ser 35 40 45Val Thr Lys Gly Ala
Met Tyr Phe His Phe Ala Ser Lys Glu Glu Leu 50 55
60Ala Arg Gly Val Leu Ala Glu Gln Thr Leu His Val Ala Val
Pro Glu65 70 75 80Ser
Gly Ser Lys Ala Gln Glu Leu Val Asp Leu Thr Met Leu Val Ala
85 90 95His Gly Met Leu His Asp Pro
Thr Leu Arg Ala Gly Thr Arg Leu Ala 100 105
110Leu Asp Gln Gly Ala Val Asp Phe Ser Asp Ala Asn Pro Phe
Gly Glu 115 120 125Trp Gly Asp Ile
Cys Ala Gln Leu Leu Ala Glu Ala Gln Glu Arg Gly 130
135 140Glu Val Leu Pro His Val Asn Pro Lys Lys Thr Gly
Asp Phe Ile Val145 150 155
160Gly Cys Phe Thr Gly Leu Gln Ala Val Ser Arg Val Thr Ser Asp Arg
165 170 175Gln Asp Leu Gly His
Arg Ile Ser Val Met Trp Asn His Val Leu Pro 180
185 190Ser Ile Val Pro Ala Ser Met Leu Thr Trp Ile Glu
Thr Gly Glu Glu 195 200 205Arg Ile
Gly Lys Val Ala Ala Ala Ala Glu Ala Ala Glu Ala Ala Glu 210
215 220Ala Ser Glu Ala Ala Ser Asp Glu225
23010276PRTStreptomyces sp. 10Met Ala Lys Gln Ala Arg Ala Val Gln
Thr Trp Arg Ser Ile Val Asp1 5 10
15Ala Ala Ala Ser Val Phe Asp Asp Tyr Gly Tyr Glu Arg Ala Ala
Ile 20 25 30Ser Glu Ile Leu
Arg Arg Ala Lys Val Thr Lys Gly Ala Leu Tyr Phe 35
40 45His Phe Ala Ser Lys Glu Ala Ile Ala Gln Ala Ile
Met Asp Glu Gln 50 55 60Thr Ser Thr
Val Glu Phe Glu Gln Glu Gly Ser Pro Leu Gln Ser Leu65 70
75 80Val Asp Gly Gly Gln Gln Phe Ala
Phe Ala Leu Arg His Asn Ser Met 85 90
95Ala Arg Ala Gly Thr Arg Leu Ser Ile Ala Gly Val Phe Leu
Gly Gly 100 105 110Pro His Pro
Trp Gly Asp Trp Ile Asp Ala Thr Ala Arg Met Leu Glu 115
120 125Leu Gly Gln Glu Arg Gly Glu Val Phe Pro Gln
Ile Asp Pro Met Val 130 135 140Ser Ala
Lys Ile Ile Val Ala Ser Phe Thr Gly Ile Gln Leu Val Ser145
150 155 160Glu Ala Asp Ser Gly Arg Ala
Asp Leu Arg Glu Gln Val Ala Glu Met 165
170 175Trp Arg His Ile Leu Pro Ser Ile Ala His Pro Gly
Val Ile Ala His 180 185 190Ile
Lys Pro Glu Gly Arg Val Asp Leu Ala Ala Gln Ala Arg Glu Lys 195
200 205Ala Glu Arg Glu Glu Gln Glu Ala Arg
Ile Ala Ala Glu Ala Lys Gly 210 215
220Ala Gly Ser Asp Pro Thr Ser Glu Gly Gly Thr Arg Ser Gly Gly Ser225
230 235 240Gly Leu Arg Gly
Gly Gly Ser Gly Arg Gly Pro Arg Ala Gly Val Thr 245
250 255Gly Asp Glu Gly Asp Glu Glu Pro Ala Gly
Ala Gly Val Ala Ala Gly 260 265
270Gly Ile Val Ala 27511214PRTStreptomyces coelicolor 11Met Thr
Ser Ala Gln Gln Pro Thr Pro Phe Ala Val Arg Ser Asn Val1 5
10 15Pro Arg Gly Pro His Pro Gln Gln
Glu Arg Ser Ile Lys Thr Arg Ala 20 25
30Gln Ile Leu Glu Ala Ala Ser Glu Ile Phe Ala Ser Arg Gly Tyr
Arg 35 40 45Gly Ala Ser Val Lys
Asp Val Ala Glu Arg Val Gly Met Thr Lys Gly 50 55
60Ala Val Tyr Phe His Phe Pro Ser Lys Glu Ser Leu Ala Ile
Ala Val65 70 75 80Val
Glu Glu His Tyr Ala Arg Trp Pro Ala Ala Met Glu Glu Ile Arg
85 90 95Ile Gln Gly Phe Thr Pro Leu
Glu Thr Val Glu Glu Met Leu His Arg 100 105
110Ala Ala Gln Ala Phe Arg Asp Asp Pro Val Met Gln Ala Gly
Ala Arg 115 120 125Leu Gln Ser Glu
Arg Ala Phe Ile Asp Ala Glu Leu Pro Leu Pro Tyr 130
135 140Val Asp Trp Thr His Leu Leu Glu Val Pro Leu Gln
Asp Ala Arg Glu145 150 155
160Ala Gly Gln Leu Arg Ala Gly Val Asp Pro Ala Ala Ala Ala Arg Ser
165 170 175Leu Val Ala Ala Phe
Phe Gly Met Gln His Val Ser Asp Asn Leu His 180
185 190Gln Arg Ala Asp Ile Met Glu Arg Trp Gln Glu Leu
Arg Glu Leu Met 195 200 205Phe Phe
Ala Leu Arg Ala 21012203PRTStreptomyces coelicolor 12Val Lys Gln Ala
Arg Ala Met Arg Thr Arg Asp Gln Val Leu Asp Ala1 5
10 15Ala Ala Glu Glu Phe Ala Leu His Gly Tyr
Ala Gly Thr Asn Leu Ala 20 25
30Thr Val Ala Val Arg Thr Gly Met Thr Lys Gly Ala Leu Tyr Gly His
35 40 45Phe Pro Ser Lys Lys Ala Leu Ala
Asp Glu Leu Val Ser Gln Ser Thr 50 55
60Glu Thr Trp Asn Thr Ile Gly Arg Ser Ile Ala Glu Thr Ala Cys Ala65
70 75 80Pro Glu Thr Ala Leu
Arg Ala Leu Val Leu Ala Val Ser Arg Gln Met 85
90 95Lys His Asp Ile Arg Phe Arg Ala Ala Leu Arg
Leu Ala Ala Asp Cys 100 105
110Thr Met Pro Ala Gly Gly Ala Pro Asp Leu Leu Asp Arg Ile Arg Arg
115 120 125Glu Met Ala Ala Ala Ala Arg
Asp Thr Gln Gln Gln Gln Ala Pro Tyr 130 135
140Ser Pro Leu Ala Thr Gln Pro Pro Asp Val Val Val His Leu Leu
Leu145 150 155 160Thr Val
Ala Tyr Gly Leu Ser Phe Ala Ala Glu Arg Gly Ala Pro Gly
165 170 175Arg Ser Pro Ala Thr Thr Asp
Lys Val Trp Glu Leu Leu Leu Thr Ala 180 185
190Leu Gln Leu Glu Asp Ile Ser Thr Cys His Asn 195
20013301PRTStreptomyces sp. 13Met Asp Ala Glu Ala Glu Val
Val His Pro Val Gly Ile Glu Met Val1 5 10
15His Arg Thr Arg Pro Glu Asp Ala Phe Pro Arg Asn Trp
Val Arg Leu 20 25 30Gly Arg
Asp Arg Phe Ala Val Glu Ala Val Leu Pro His Asp His Pro 35
40 45Phe Phe Ala Pro Val Gly Asp Asp Leu His
Asp Pro Leu Leu Val Ala 50 55 60Glu
Ala Met Arg Gln Ala Ala Met Leu Ala Phe His Ala Gly Tyr Gly65
70 75 80Ile Pro Leu Gly Tyr His
Phe Leu Leu Thr Glu Leu Asp Tyr Val Cys 85
90 95His Pro Glu His Leu Gly Val Gly Gly Glu Pro Thr
Glu Ile Gly Leu 100 105 110Glu
Val Phe Cys Ser Asp Leu Lys Trp Arg Ala Gly Leu Pro Ala Gln 115
120 125Gly Arg Val Gly Trp Ala Val His Arg
Gly Asp Arg Leu Ala Ala Thr 130 135
140Gly Val Ala Ala Thr Arg Phe Ser Thr Pro Lys Ala Tyr Arg Arg Met145
150 155 160Arg Gly Asp Val
Pro Val Glu Gly Ile Ser Leu Pro Glu Thr Ala Pro 165
170 175Val Pro Ala Ser Pro Ala Gly Arg Ala Arg
Val Glu Asp Val Val Leu 180 185
190Ser Gly Thr Gly Arg Glu Gly Val Trp Glu Leu Arg Val Asp Thr Arg
195 200 205His Pro Thr Leu Phe Gln Arg
Pro Asn Asp His Val Pro Gly Met Leu 210 215
220Leu Leu Glu Ala Ala Arg Gln Ala Ala Cys Leu Val Ala Gly Pro
Ala225 230 235 240Gly Ile
Val Pro Val Glu Ala Arg Thr Arg Phe His Arg Tyr Ser Glu
245 250 255Phe Gly Ser Pro Cys Trp Ile
Gly Ala Val Val Gln Pro Gly Ala Asp 260 265
270Glu Asp Thr Val Thr Val Arg Val Thr Gly His Gln Asp Gly
Glu Thr 275 280 285Val Phe Ser Thr
Val Leu Ser Gly Pro Arg Ala His Gly 290 295
30014314PRTStreptomyces sp. 14Met Pro Glu Ala Val Val Leu Ile Asn
Ser Ala Ser Asp Ala Asn Ser1 5 10
15Ile Glu Gln Thr Ala Leu Pro Val Pro Met Ala Leu Val His Arg
Thr 20 25 30Arg Val Gln Asp
Ala Phe Pro Val Ser Trp Ile Pro Lys Gly Gly Asp 35
40 45Arg Phe Ser Val Thr Ala Val Leu Pro His Asp His
Pro Phe Phe Ala 50 55 60Pro Val His
Gly Asp Arg His Asp Pro Leu Leu Ile Ala Glu Thr Leu65 70
75 80Arg Gln Ala Ala Met Leu Val Phe
His Ala Gly Tyr Gly Val Pro Val 85 90
95Gly Tyr His Phe Leu Met Ala Thr Leu Asp Tyr Thr Cys His
Leu Asp 100 105 110His Leu Gly
Val Ser Gly Glu Val Ala Glu Leu Glu Val Glu Val Ala 115
120 125Cys Ser Gln Leu Lys Phe Arg Gly Gly Gln Pro
Val Gln Gly Gln Val 130 135 140Asp Trp
Ala Val Arg Arg Ala Gly Arg Leu Ala Ala Thr Gly Thr Ala145
150 155 160Thr Thr Arg Phe Thr Ser Pro
Gln Val Tyr Arg Arg Met Arg Gly Asp 165
170 175Phe Ala Thr Pro Thr Ala Ser Val Pro Gly Thr Ala
Pro Val Pro Ala 180 185 190Ala
Arg Ala Gly Arg Thr Arg Asp Glu Asp Val Val Leu Ser Ala Ser 195
200 205Ser Gln Gln Asp Thr Trp Arg Leu Arg
Val Asp Thr Ser His Pro Thr 210 215
220Leu Phe Gln Arg Pro Asn Asp His Val Pro Gly Met Leu Leu Leu Glu225
230 235 240Ala Ala Arg Gln
Ala Ala Cys Leu Val Thr Gly Pro Ala Pro Phe Val 245
250 255Pro Ser Ile Gly Gly Thr Arg Phe Val Arg
Tyr Ala Glu Phe Asp Ser 260 265
270Pro Cys Trp Ile Gln Ala Thr Val Arg Pro Gly Pro Ala Ala Gly Leu
275 280 285Thr Thr Val Arg Val Thr Gly
His Gln Asp Gly Ser Leu Val Phe Leu 290 295
300Thr Thr Leu Ser Gly Pro Ala Phe Ser Gly305
31015314PRTStreptomyces sp. 15Val Ala Val Pro Ala Arg Arg Thr Ala Phe Gln
Thr Gly Arg Pro Ala1 5 10
15Arg Ala Ser Ala Met Thr Ser Thr Val Pro Arg Glu Leu Val His Arg
20 25 30Ala Ala Val Ala Glu Val Phe
Leu Thr Gly Trp Ser Arg Thr Ala Glu 35 40
45Asn Arg Phe Ala Leu Thr Ala Gln Trp Pro Arg Ala His Ser Tyr
Phe 50 55 60Thr Pro Val Asn Gly Cys
Tyr Asp Pro Leu Leu Ala Ser Glu Thr Ile65 70
75 80Arg Gln Val Gly Thr Leu Leu Ser His Ala Glu
Phe Gly Val Ser Phe 85 90
95Gly Asp Gln Phe Leu Met Trp Asp Leu His His Ser Val Arg Pro Glu
100 105 110Gln Ala Gly Val Gly Ala
Ala Pro Ala Asp Leu Glu Leu Asp Val Ile 115 120
125Cys Ser Asp Ile Arg Arg Arg Gly Arg Arg Leu Ala Gly Met
Arg Tyr 130 135 140Glu Val Thr Leu Tyr
Cys Gly Gly Gln Val Ile Ala Thr Gly Gly Ala145 150
155 160Ala Phe Asp Cys Thr Ser Pro Ala Val Tyr
Gln Arg Leu Arg Gly Asp 165 170
175Arg Val Gly Ala Thr Gly Val Arg Pro Leu Pro Gln Pro Leu Ala Pro
180 185 190Ala Ser Val Gly Arg
Phe Leu Thr Thr Asp Val Val Leu Ser Ala Thr 195
200 205Glu Arg Pro Leu Glu Trp Gln Leu Arg Val Asp Glu
Gln His Pro Val 210 215 220Leu Phe Asp
His Pro Val Asp His Val Pro Gly Met Val Leu Met Glu225
230 235 240Ser Ala Arg Gln Ala Ala Gln
Ala Ile Asp Pro Ser Arg Pro Phe Leu 245
250 255Pro Thr Thr Met Arg Ser Glu Phe Ser Arg Tyr Ala
Glu Leu Asp Arg 260 265 270Pro
Cys Trp Ile Gln Ala Glu Pro Leu Pro Ala Ala Asp Asn Gly Asp 275
280 285Arg Gln Val Arg Val Thr Gly His Gln
Asp Asp Thr Thr Val Phe Ser 290 295
300Cys Leu Ile Gly Thr Arg Gly Ala Ala Glu305
31016291PRTStreptomyces sp. 16Leu Val His Arg Thr Ser Thr Ala Gln Val Leu
Leu Thr Asp Trp Gln1 5 10
15Arg Leu Asp Asp Ala Arg Phe Ser Val Thr Ala Arg Trp Pro Leu Ser
20 25 30His Ala Phe Phe Thr Pro Val
Gly Asp Gly Tyr Tyr Asp Pro Leu Met 35 40
45Cys Ala Glu Thr Ile Arg Gln Ile Ala Tyr Leu Leu Gly His Ala
Glu 50 55 60Phe Ala Val Pro Phe Gly
His Gln Phe Val Leu Trp Asp Leu Ser Val65 70
75 80Ser Val Val Arg Pro Glu Leu Leu Arg Val Gly
Leu Val Pro Ala Thr 85 90
95Val Asp Leu Ala Ile Thr Cys Val Glu Ile Lys Arg Arg Ala Gly Arg
100 105 110Leu Ser Gly Leu Gly Tyr
Glu Ala Val Val Arg Arg Asp Gly Gln Val 115 120
125Val Ala Thr Gly Arg Ala Ser Val Thr Cys Thr Ser Pro Ala
Val Tyr 130 135 140Gln Arg Ile Arg Pro
Glu His Val Leu Thr Pro Glu His Arg Pro Leu145 150
155 160Pro Leu Thr Ala Pro Ala Ala Pro Gln Ser
Val Ala Arg Leu Ser Pro 165 170
175Thr Asp Val Val Leu Ser Pro Leu Asp Arg Glu Asn Arg Trp Gln Leu
180 185 190Arg Val Asp Thr Asn
His Pro Val Leu Phe Asp His Trp Val Asp His 195
200 205Val Pro Gly Met Val Leu Met Glu Ala Ala Arg Gln
Ala Ala Ala Ser 210 215 220Ala Leu Gly
Arg Pro Ser Phe Met Pro Leu Gly Val Ala Gly Glu Phe225
230 235 240Lys Arg Tyr Val Glu Leu Asp
Ala Pro Cys Val Ile Glu Ser Glu Arg 245
250 255Leu Phe Gln Asp Val Pro Gly Ala Glu Glu Val Val
Arg Val Thr Gly 260 265 270His
Gln Asn Gly Glu Leu Thr Phe Val Gly Thr Val Thr Ala Ser Ser 275
280 285Tyr Gly Tyr
29017353PRTStreptomyces coelicolor 17Met Asn His Thr Asn Arg Leu Leu Leu
Pro Ala Pro His Asp Leu Leu1 5 10
15Phe Asp Gly Cys Pro Pro Leu Ser Phe Ala Arg Pro Leu Pro Pro
Ala 20 25 30Asp Val His Lys
Ala Ala Ala Ala Glu Val Leu Leu Thr Asp Ala Arg 35
40 45Pro Leu Gly Glu Asn Arg Phe Ala Val Ala Ala Leu
Trp Pro Arg Asn 50 55 60Thr Phe Leu
Ala His Arg Ala Thr Ser Ser Pro Cys Asp Pro Leu Leu65 70
75 80Ala Ala Glu Thr Ile Arg Gln Ser
Ala Ile His Leu Ser His Thr Phe 85 90
95Cys Asp Val Pro Ile Gly His His Phe Val Leu Ser Gly Leu
Asp Leu 100 105 110Asp Leu Asp
Leu Pro Val Trp Asp Ser Gly Pro Leu Pro Val Val Leu 115
120 125Asp Val Thr Ser Thr Lys Thr Thr Thr Asn Pro
Arg Arg Met Ala Arg 130 135 140Ala Leu
Asn Ala Asp Val Tyr Val Ala Gly Leu His Arg Gly Arg Cys145
150 155 160Ala Ile Arg Phe Glu Val Leu
Ala Pro Arg Arg Tyr Ala Met Ile Arg 165
170 175Asp Arg Ala Arg Arg Ala Glu Arg Pro Ala Gln Gln
Ala Ala Ala Gly 180 185 190Ala
Ala Thr Ala Leu Pro Pro Glu Thr Val Gly Phe His Asp Asp Leu 195
200 205His Val Leu Leu Ala Thr Ala Gln Gly
Leu Pro Asp Thr Ala Trp Gln 210 215
220Leu Arg Leu Arg Arg Asp His Pro Val Leu Phe Asp His Glu Ser Asp225
230 235 240His Ile Ser Gly
Met Ala Leu Leu Glu Ala Cys Arg Gln Ala Ala Thr 245
250 255Ala Leu Thr Pro Pro Ala Pro Gly Ala Phe
Gly Pro Arg Gln Val Ala 260 265
270Leu Thr Ala Val Ala Ser Ser Tyr Gln Ala Phe Gly Glu Leu Asp Ser
275 280 285Pro Val Thr Ile Thr Thr Leu
Pro Ala Ala His Gly His Ser Pro Asp 290 295
300Ser Gly Thr Arg Thr Leu Gln Leu Thr Ala Arg Gln Gly Ser Arg
Thr305 310 315 320Leu Ile
Thr Ala Thr Val Thr Thr Thr Thr Thr Ala Gly Thr Gly Ser
325 330 335Pro Gly Pro Thr Val Pro His
His Gly Asp Gln Thr Lys Ala Val Ala 340 345
350Ser189521DNAStreptomyces coelicolor 18ctcgagacat
ggctggcatc gaggtcgacg acaccactgc ggacgagctg cgggccctgg 60ccgacgcggc
cgggctgccg ctggacgcct acctcgcgca ggtcgccgag gagaagcggc 120gcgagcgcgc
gctggccgag ggcgcggaga tcttccgccg ggtcaccggc accccggaga 180ccgtcgccgc
cttcgacgcg gagtacggcg gccccgcgca ggcgcagacc gccccgcggg 240cggcctgacc
tgtgcctgcc gagtactacg tcgactaccg gtggttcctg gagcgccagg 300ccgagctgct
ggacgatctc gcggtcagcg actactccgt cttcgtcggc ctagccgccc 360ggcacagggt
cgacccgccc cgtcacgacc agcatcaccc ggacgccttc tggcgggcgg 420ccgtgatgct
ggaggagtgc gtcgtgctcc ggcccctgcc cgcccgcaac gagctgtacg 480gcttcggcgt
ggccgtggcg tacctcggga tgcacgggga gcgggtgaac acgaaaattc 540gaggcctggc
gggacctgat ctccgacatc accgccctgc gtctcgactc cttcgccgtc 600gccgagcggc
tgcgctcgct ccgtctgccg ccggcctgac ctcgctgtcg ctctcccccg 660caggaaacgg
actgcctgat ccgtaaccgg acgccacgcg ctctggcgct cttctttgcc 720ggtgcccgtg
agcccggagc cggcatggtc tgcctgggcc ggagctcgtt gtccgccgga 780tcctggtcgc
cgcagtcagt tgtggcaggt tgaaatgtcc tccaactgca gcgcagtaag 840caggagttcc
cacaccttgt cagtcgtggc aggtgagcgt cccggcgccc ctcgttcagc 900cgcgaacgac
aacccatagg caacggtcag cagcaggtgg accaccacgt ccggtggctg 960cgtggccaag
ggtgagtagg gggcttgctg ttgctgggtg tcacgggcgg ccgcggccat 1020ctcgcggcgg
atgcggtcca ggagatccgg cgccccgccc gcgggcatgg tgcagtccgc 1080tgccagccgc
agcgccgcac ggaaccggat gtcgtgcttc atttgacggc ttacggccag 1140cacgagggca
cgcaaggcgg tttcgggcgc gcaggcggtc tcggcgatgg agcggccgat 1200ggtgttccat
gtctctgtcg actggctcac cagttcgtcg gcgagcgcct tcttggatgg 1260gaagtgcccg
tagagagcgc ccttcgtcat gcctgtgcgt acggcgaccg ttgccagatt 1320ggtgcctgca
tagccgtgca gggcgaactc ttcagccgcc gcatccagca cctggtcccg 1380ggtgcgcatc
gcccttgcct gcttcaccaa cgcccgagtc ctctcaaggt cgtgagccaa 1440cgggcccgga
aaacataccc tcgggaaggt atgttagtgg gggcggtcgg cgcacgtgga 1500cgatgtccca
gtcatcggcg caaaagtgga ggcagcacgg ctaccgcgcg gcaggattca 1560accgatggcc
gacggatcgt cgtggcctag cgggcctgga cgccgtgcgc atgcacagcg 1620gggtcccgcg
tcggcgggca gcaccggcca gttggacccc tgggcgtggc gcaccagcac 1680gaggtccgtg
ccgaccacga ccgggttccc cacggcctgc agcatgccga agtcgctctc 1740gtggtccccg
taggcaaagc agtctgccgg caccaccccc ctcttcgcca tcacttcggt 1800cacggcctca
gccttcgctt cgccgatcat cgggcgattc acctcgccgg tgaggacgcc 1860ctgggcgtcg
gcgaactgct cggtgcacag aatccggtcc gcgccgaggt cctgcgccag 1920gggcgtgagc
agtggccggg ccgagcccga gatcagaacg atcgtgtggc cggcccggcg 1980gtgccgagcg
agtgccgcca ggccggccct gacgtagccg tccggccgcg tgcggtaagc 2040gtggtaccag
tcgcggccgg cctcctgcag gcgagccagg gaaacaccgg cgtagcgccg 2100gtagtagacg
cggttcatct ccacccggct cgcccctcgg cgccgcatcg ccgtcagatc 2160ggcatcagca
ctgtggcgtt gcccgctcgc ctgcgcggtg atgtcgtccc gcaagctgtg 2220cggcgcctgc
cgtgcgaagt cgagcatgct cttggcggtg atcagtgtct cgtccacatc 2280gaagaaggcg
atggggcgta tggcgggcga ccggttcgcg gctgtgcgac gttcgcgcgg 2340gggctcgggc
atcacgccgt acgtctttct gtgtctggtg cggtcgcggt tctgccgggt 2400gggccggccg
ggccgcccgg ctcggcgggg ccgacgccgg tcgatgggtc cgcacggtcg 2460aagagagcag
gcgtgtatgc gccggctgct gcgtcgatgc tcagggccct gtgcgtggca 2520gcggtggtga
tgtcccgcca gtggcgctgt accgggtcgt cttctgcctg gccacgtgat 2580cccgaggcgc
gcagcagttg gtcgacggct tcggagcaca gttccacagc cgcggcggcg 2640tcccgctgcc
cctcggcgac gaggagcggg gtcactggcg cgtggtcggc ccgctctgcc 2700gctgcctcca
ggagcaggcc tgcggcgcgt atgcgtgctg ctgctctggt cagggtgttg 2760gacgccgggg
gcactgcggt gccctgtcgt tctgtggcgg cgtgtgtcca ggcgtcaaga 2820gctccgcggg
ccgccccgag aaccggaaag gcgaacatca gcgcgcccac catggcgtag 2880ggcaccgtgt
ggcagcgggc cgagccgggc agggggagca gcaggtccga caaggtgcag 2940gtgcggtggc
ggggaaccag caccccgtcc gcctcgacgg tgttgctgcc ggtcccgcgc 3000atgccgaggg
tgtgccaggt gtcggtgacc gtcagctcgt ccctggggac ggcgaacagc 3060cggtgccgct
cgggaacgtt ccggcccggt gtccagcttg cgagcagcac ccagtcggcg 3120tggtcgacgc
cgctggcgaa tccccagcgc ccggtgagcc gccagccgcc cggctcgagg 3180ttggcctcgc
ccgacggggg catgatggcc gcggcgatac gggcgtcggg cgaggagtgc 3240cacagttcgc
gttgggcctt ttcgggcagg tacgaggcca gccgcccatg ggccgcatac 3300agcgtggcgc
accaggcggt ggcggcgcag gtccgggcga gcgtggtcgc cgccgtgagc 3360agttcgccga
aggtcccggc gcggccgccg aagcgccggg ggacgaagtg gcgtggaaag 3420ccgacgtcgg
tgaccgcccg ggccacgtcg tctgtgagtc gtcggtgtgt ctcctggact 3480ccgtggtccc
ggtgcgcgag agccacggcg tgttccaccc cgtcgcggga aaactccctg 3540agcggcgcgg
tcatgaggcc accgccttcg tctggtcgcc gtggtgcggg acggtgggcc 3600cggggcttcc
cgtcccggct gtcgtcgtcg tggtgacggt cgcggtgatg agcgtgcggc 3660tgccttgccg
ggcggtgagt tgcagcgtac gtgtgccgct gtccgggctg tgcccgtggg 3720cggcggggag
ggtggtgatg gtgacagggg agtcgagttc gccgaatgcc tggtaggaac 3780ttgcgacggc
cgtgagggcc acctgccgcg ggccgaaggc tccgggcgcg ggtggggtga 3840gggctgtggc
ggcctgacgg caggcctcca gcagtgccat gccggaaatg tggtccgatt 3900cgtggtcgaa
gaggaccgga tggtcccggc gcagccgcag ttgccaggct gtatcgggca 3960ggccttgcgc
ggtggcaagc aggacgtgca ggtcgtcgtg gaagccgacg gtttcgggag 4020gaagggcggt
cgccgcgcct gcggctgcct gctgtgcggg gcgctctgcc cgcctggcgc 4080gatcgcggat
catcgcgtac cgccgggggg cgaggacctc gaagcggatg gcgcagcggc 4140cgcggtggag
tccggccacg tacacgtcgg cgttcaacgc cctggccatc cggcgcgggt 4200tcgtggtggt
cttcgtactc gtgacgtcca ggacgacagg cagcgggccg gagtcccaga 4260cagggagatc
gagatcaaga tcgaggcccg acagcacgaa gtggtggcct atgggcacgt 4320cacagaaggt
gtgtgagagg tggatcgccg actgtcgtat ggtctccgcg gctaggaggg 4380ggtcgcacgg
gctcgatgtc gcgcggtgcg cgaggaaggt gtttcggggc cacagggcgg 4440cgacggcgaa
ccggttctcg cccagcggtc gcgcgtcggt gaggagtact tctgccgcgg 4500cagccttgtg
tacgtcggcc ggcggcaggg ggcgcgcgaa ggagagcgga gggcagccgt 4560cgaacaggag
gtcgtggggg gcgggcagta aaagacggtt tgtatggttc ataggggcgc 4620tacatctccc
ggtgtgtcct cgtacgggac caccggctgg cttgccgcgc tgcaagacag 4680ccgggatcgg
taagctgacc gagagaaata tacctgcggg aaggtattat gcaatgggtt 4740tccgtgccga
cccgggtcgc accagcatgg cgcccgcagg gcccgcacac acgaaggaag 4800gcagccatga
cgagcgccca acaacccacg cctttcgcgg tccggtccaa cgtgccgcgt 4860ggacctcacc
cgcagcagga gcggtcgatc aagacccggg cccagatcct ggaggcggcg 4920tcggagatct
tcgcgtcgcg cggctaccga ggggcctccg tcaaggacgt tgccgagcgt 4980gtcggcatga
ccaagggcgc ggtgtacttc cacttcccca gcaaggaatc actggccatc 5040gccgtggtgg
aggagcacta cgcgcgctgg cccgcagcga tggaagagat ccgcatccag 5100ggcttcacac
cgctggagac ggtcgaggag atgctccatc gcgcggcgca ggccttccgc 5160gacgaccccg
tgatgcaggc cggtgcccgg ctgcagagtg agcgcgcctt catcgacgcg 5220gagctgcccc
tgccctacgt ggactggacc cacctgctgg aggtgccgtt gcaggacgcc 5280cgtgaggccg
gccagttgcg ggcgggcgtc gatcccgcag cagctgcccg ttccctggtg 5340gccgccttct
tcggcatgca gcacgtctcc gacaatctgc accagcgagc ggacatcatg 5400gagcggtggc
aggagctgcg ggagctgatg ttcttcgctc tccgcgcctg acggggagcg 5460tccgcaaaac
tggtggtgcc actgatagga gaatctccct cttttccctg cgctccagca 5520ccgattacgt
tctctgcatg attgcggaca ccgcgacgac cagcgcgcgg acgggagccc 5580cggccgcagc
gttgcgtctg ttctgttttc atcatgcagg aggccaggga acagcattcc 5640tcggatggca
gaagagactg ggagcgcggg cggaggtgat tcccgtccgg ctgcccccgc 5700ccgaggacgt
ctctgcagag acagcggacg ggagcggaat gtcgatgacc ctcgtagtcg 5760cttccctcga
tcacgaactc ggcccaatgc tgcggcggcc cttcctgttc tacgggcaca 5820gcatgggcgc
tctcgtggcc taccacctca cccgcctgcg ccagtcccgc ggccggcccc 5880tgccggagcg
gttgctcatc ggcgcctacc cggcccccca tctgccgcac cggctcgccc 5940actgcacgca
cttgcctgac gaggacctgc tcgcgctgct gccgccgcac cctgccggcc 6000actctcgcct
gctgcgccag gcgcccggcc tggcgacagc gactgcggcg cggctgcgcc 6060tgcacctcgg
cctgtgtgac agcgccgcgc cggcggcacc gaaccccgcg cagcacaccg 6120gccacggttc
cccgcagggg aggagtgaac cgctgaggtg tccggtggat gtgttcaccg 6180ggatcagcga
tccgctggtg acggacgccg aggcagccgc atggcggcac cacacccgcg 6240caggctgccg
tatacaccgc atccccggcg ggcatttctt cacgcgcgag accccggaat 6300ctagggccgc
gttcttcgac cggctgtgca cggtgcttgc agggccgtcg gaatgggcgg 6360ccggagcatc
gggtcccctc cctgtcaccg tcgcttcgta aaagcgtttc ccgcaaccca 6420ggaggacgtt
catgtacccc gagacgctcg gattcggtgc tttcctctcc cccatgcatc 6480cgctgggcga
gaatcccacg ctgcaatttc agcgcgacct tgagctgata gaactcctcg 6540accggctcga
ctacaacgaa ttctgggtcg gcgagcatca ctccatgggc tggaacacca 6600tcggcagccc
ggagctgatg gttgcggctg ccgccgagcg gacccgtcgt atcaccctgg 6660ccaccggtgt
gatgacgctg ccgtaccacc acccgttcat ggtggcgagc cgtgcggtgc 6720acctcgacca
tctgacccgt ggccggttcg tgctcggtgt gggcgcgggc ggcatcccga 6780ccgacgcccg
catgatcggc cgtgagatga gcgaactgcg caccatgttc ggcgaggcac 6840tggaggcggt
cgtcgcgctg gtcaacggcg aggagcgggt gaccaagaag acctcgtggt 6900tcacgctgaa
ggacgccaag ctccagctgt ccccgtaccg tgcatcaggg ctggagatcg 6960ccgctgccag
cgtcgcctcc ggcaacagca tgcggctggc cggccgctac gggatcagca 7020ccgtctcctt
cggtgcgccg cggcctggtc atccccgacc cgacatgcgt acccagtggt 7080cgtatgcgga
ggaggctgcg gccgaacagg gcaccacggt ggaccgcagg aactggcgaa 7140tcaccctgcc
ggtatacgtg gcagagacgc gcgagcaggc ccttgccgat gtccgggagg 7200gttacgaccg
ctgggcctac ggatactggg gcgacatccg cggcctcgac gtcagcgtcc 7260ccggcgtcaa
gcgtgcgcag gctctggagg ctgccgtgga cgcgggcagc gccatcgtcg 7320gctccgtcga
ggacgtggtg gccggcgtcg agcggctccg tgaggaggtc ggcggcttcg 7380ggaccctgct
cgtctacgcg caggactggg ccgactggga gaagacgaag cggagctatg 7440acctgctggc
ccgctacgtc gccccgcact tcaccggctc cacccggcga ctgtacgagt 7500cggtgcagtg
gtaccaggac aaccgcgacc tgtttccgca gctcatcccg taaaccgtgc 7560acgccgtgcc
tgccggcgcc acgggcagct ccaggcacgg ctcggcatcc ctccctggag 7620tgacagcgac
accccatggc caccgaaccg atacgcatcg gcgtggtcgg cgcctccccg 7680gaccggggct
gggccgccga cgcacacctg ccggccctgc agcacctgcc gcagtacaag 7740atcaccgcgg
tcggcacccg ccgggcggac agtgcgcacc gggccgctcg ccggtacggg 7800gcgacccacg
ctttcaccga cccccgcagc ctcgccgcac atcccgacgt ggaactggtc 7860gcgatcgtcg
tgaaagtgcc ggaccatgcg cggctggtgg aggcggcgct cgcggcgggc 7920aagcatgtcc
tgtgcgagtg gccccttgcc cggaccaccg aggaggccgc ccagctaacg 7980gcggccgctc
acggagccgg tgtggtgaac gccgtcggcc tccaggcgcg gcacaccccg 8040acggtcgtcc
gggcccggga actgatcagg caggggtacg tcggccgggt cacctcggtc 8100accgtgtaca
gcacgcgggg ggtcgcggcc ggggggcggc tgcccgccgc cttcgcctac 8160accctcgact
ccacgaacgg cgccggcacc ttcgaggtcg ccggcgggca cacgctcgac 8220gcggtgcagt
acctgctcgg cagggagatg accggcctgt cggctgcgct gtccgttcag 8280catccgcgga
tcacactcga cgaggacgcc cggcagacgg gggcgaccag ccccgatcat 8340gtcgcgctgc
acgcgacgct ggaaggcggc gccgcgctgg tggtccacat ccacgatgcc 8400aagaacagcg
gcgcgggcac ccgcatcgag atctccggca cgcaagggga gctggccatc 8460gtatccaccg
gaccacgaag cggcagcggg ctgcagatca gcgaactggc cctgctcgga 8520gcgcagggga
cagagccgtc cgggcaggag ctgccctttc ccggctcctg gggcacggcc 8580gtgccagcgg
acggtctcga tgcggcccag cacaccatgg ctgtgcagta cgcggctctg 8640gccgcggaca
tccgcgaggg cggcagtcgt gtgcctcgtt tcgccgacgg gatcgagctg 8700caccggctgc
tggacgccgt acggctgtcc tccgcaaccg gctgccggct ggagcgccgt 8760gcgggcgagc
ggtggccggt cagctctccc tggccgcggc gtcgacgatc gcggtgagca 8820ggccggggaa
ggcctgatcg agatcatcgg tgcgaagcgt gttcatcttc gaggtgccga 8880tgtagtactg
cctgatgatc ccggcctggc gcaacacctt gaagtggtga gtgccggtcg 8940agcgggagac
ggtgatgtcg aaggtgccgc aggcgatgtc ctcgggtgcc ttagccagct 9000gccggacgat
gctgcggcgc accggatcga ccagcgcgtc caggacgccc tggagggtga 9060tggcgtcagc
gtccggatgg tcggtgatgc gctctgtcgt gatccgtgcc gccacggctg 9120tccgcctcct
cgtcgcgtgt cgttccacca tggtacgaca gccatcaaat gttgacggcc 9180atcaaagttt
gacagccgtc gtcatatgag cttcagtgag aacgcacggt aattccggcg 9240cagttgggcg
gccgccatcc ccccccggcc gccctctgcc ggccactcct caggacgcgg 9300cccatacccg
acccccacag gagcagaaca gcatgaccac tgtccgaaca ggcggggcgc 9360agaccgccga
agtcccggcg ggcggccggc gcgatgtccc cagcggggtg aagatcaccg 9420ctctggccac
gggattcgtc atggcgaccc tggacgtcac cgtggtgaac gtcgccggag 9480ccaccatcca
ggagagcctg gacaccacgc tgacccagct g
9521199521DNAStreptomyces coelicolor 19cagctgggtc agcgtggtgt ccaggctctc
ctggatggtg gctccggcga cgttcaccac 60ggtgacgtcc agggtcgcca tgacgaatcc
cgtggccaga gcggtgatct tcaccccgct 120ggggacatcg cgccggccgc ccgccgggac
ttcggcggtc tgcgccccgc ctgttcggac 180agtggtcatg ctgttctgct cctgtggggg
tcgggtatgg gccgcgtcct gaggagtggc 240cggcagaggg cggccggggg gggatggcgg
ccgcccaact gcgccggaat taccgtgcgt 300tctcactgaa gctcatatga cgacggctgt
caaactttga tggccgtcaa catttgatgg 360ctgtcgtacc atggtggaac gacacgcgac
gaggaggcgg acagccgtgg cggcacggat 420cacgacagag cgcatcaccg accatccgga
cgctgacgcc atcaccctcc agggcgtcct 480ggacgcgctg gtcgatccgg tgcgccgcag
catcgtccgg cagctggcta aggcacccga 540ggacatcgcc tgcggcacct tcgacatcac
cgtctcccgc tcgaccggca ctcaccactt 600caaggtgttg cgccaggccg ggatcatcag
gcagtactac atcggcacct cgaagatgaa 660cacgcttcgc accgatgatc tcgatcaggc
cttccccggc ctgctcaccg cgatcgtcga 720cgccgcggcc agggagagct gaccggccac
cgctcgcccg cacggcgctc cagccggcag 780ccggttgcgg aggacagccg tacggcgtcc
agcagccggt gcagctcgat cccgtcggcg 840aaacgaggca cacgactgcc gccctcgcgg
atgtccgcgg ccagagccgc gtactgcaca 900gccatggtgt gctgggccgc atcgagaccg
tccgctggca cggccgtgcc ccaggagccg 960ggaaagggca gctcctgccc ggacggctct
gtcccctgcg ctccgagcag ggccagttcg 1020ctgatctgca gcccgctgcc gcttcgtggt
ccggtggata cgatggccag ctccccttgc 1080gtgccggaga tctcgatgcg ggtgcccgcg
ccgctgttct tggcatcgtg gatgtggacc 1140accagcgcgg cgccgccttc cagcgtcgcg
tgcagcgcga catgatcggg gctggtcgcc 1200cccgtctgcc gggcgtcctc gtcgagtgtg
atccgcggat gctgaacgga cagcgcagcc 1260gacaggccgg tcatctccct gccgagcagg
tactgcaccg cgtcgagcgt gtgcccgccg 1320gcgacctcga aggtgccggc gccgttcgtg
gagtcgaggg tgtaggcgaa ggcggcgggc 1380agccgccccc cggccgcgac cccccgcgtg
ctgtacacgg tgaccgaggt gacccggccg 1440acgtacccct gcctgatcag ttcccgggcc
cggacgaccg tcggggtgtg ccgcgcctgg 1500aggccgacgg cgttcaccac accggctccg
tgagcggccg ccgttagctg ggcggcctcc 1560tcggtggtcc gggcaagggg ccactcgcac
aggacatgct tgcccgccgc gagcgccgcc 1620tccaccagcc gcgcatggtc cggcactttc
acgacgatcg cgaccagttc cacgtcggga 1680tgtgcggcga ggctgcgggg gtcggtgaaa
gcgtgggtcg ccccgtaccg gcgagcggcc 1740cggtgcgcac tgtccgcccg gcgggtgccg
accgcggtga tcttgtactg cggcaggtgc 1800tgcagggccg gcaggtgtgc gtcggcggcc
cagccccggt ccggggaggc gccgaccacg 1860ccgatgcgta tcggttcggt ggccatgggg
tgtcgctgtc actccaggga gggatgccga 1920gccgtgcctg gagctgcccg tggcgccggc
aggcacggcg tgcacggttt acgggatgag 1980ctgcggaaac aggtcgcggt tgtcctggta
ccactgcacc gactcgtaca gtcgccgggt 2040ggagccggtg aagtgcgggg cgacgtagcg
ggccagcagg tcatagctcc gcttcgtctt 2100ctcccagtcg gcccagtcct gcgcgtagac
gagcagggtc ccgaagccgc cgacctcctc 2160acggagccgc tcgacgccgg ccaccacgtc
ctcgacggag ccgacgatgg cgctgcccgc 2220gtccacggca gcctccagag cctgcgcacg
cttgacgccg gggacgctga cgtcgaggcc 2280gcggatgtcg ccccagtatc cgtaggccca
gcggtcgtaa ccctcccgga catcggcaag 2340ggcctgctcg cgcgtctctg ccacgtatac
cggcagggtg attcgccagt tcctgcggtc 2400caccgtggtg ccctgttcgg ccgcagcctc
ctccgcatac gaccactggg tacgcatgtc 2460gggtcgggga tgaccaggcc gcggcgcacc
gaaggagacg gtgctgatcc cgtagcggcc 2520ggccagccgc atgctgttgc cggaggcgac
gctggcagcg gcgatctcca gccctgatgc 2580acggtacggg gacagctgga gcttggcgtc
cttcagcgtg aaccacgagg tcttcttggt 2640cacccgctcc tcgccgttga ccagcgcgac
gaccgcctcc agtgcctcgc cgaacatggt 2700gcgcagttcg ctcatctcac ggccgatcat
gcgggcgtcg gtcgggatgc cgcccgcgcc 2760cacaccgagc acgaaccggc cacgggtcag
atggtcgagg tgcaccgcac ggctcgccac 2820catgaacggg tggtggtacg gcagcgtcat
cacaccggtg gccagggtga tacgacgggt 2880ccgctcggcg gcagccgcaa ccatcagctc
cgggctgccg atggtgttcc agcccatgga 2940gtgatgctcg ccgacccaga attcgttgta
gtcgagccgg tcgaggagtt ctatcagctc 3000aaggtcgcgc tgaaattgca gcgtgggatt
ctcgcccagc ggatgcatgg gggagaggaa 3060agcaccgaat ccgagcgtct cggggtacat
gaacgtcctc ctgggttgcg ggaaacgctt 3120ttacgaagcg acggtgacag ggaggggacc
cgatgctccg gccgcccatt ccgacggccc 3180tgcaagcacc gtgcacagcc ggtcgaagaa
cgcggcccta gattccgggg tctcgcgcgt 3240gaagaaatgc ccgccgggga tgcggtgtat
acggcagcct gcgcgggtgt ggtgccgcca 3300tgcggctgcc tcggcgtccg tcaccagcgg
atcgctgatc ccggtgaaca catccaccgg 3360acacctcagc ggttcactcc tcccctgcgg
ggaaccgtgg ccggtgtgct gcgcggggtt 3420cggtgccgcc ggcgcggcgc tgtcacacag
gccgaggtgc aggcgcagcc gcgccgcagt 3480cgctgtcgcc aggccgggcg cctggcgcag
caggcgagag tggccggcag ggtgcggcgg 3540cagcagcgcg agcaggtcct cgtcaggcaa
gtgcgtgcag tgggcgagcc ggtgcggcag 3600atggggggcc gggtaggcgc cgatgagcaa
ccgctccggc aggggccggc cgcgggactg 3660gcgcaggcgg gtgaggtggt aggccacgag
agcgcccatg ctgtgcccgt agaacaggaa 3720gggccgccgc agcattgggc cgagttcgtg
atcgagggaa gcgactacga gggtcatcga 3780cattccgctc ccgtccgctg tctctgcaga
gacgtcctcg ggcgggggca gccggacggg 3840aatcacctcc gcccgcgctc ccagtctctt
ctgccatccg aggaatgctg ttccctggcc 3900tcctgcatga tgaaaacaga acagacgcaa
cgctgcggcc ggggctcccg tccgcgcgct 3960ggtcgtcgcg gtgtccgcaa tcatgcagag
aacgtaatcg gtgctggagc gcagggaaaa 4020gagggagatt ctcctatcag tggcaccacc
agttttgcgg acgctccccg tcaggcgcgg 4080agagcgaaga acatcagctc ccgcagctcc
tgccaccgct ccatgatgtc cgctcgctgg 4140tgcagattgt cggagacgtg ctgcatgccg
aagaaggcgg ccaccaggga acgggcagct 4200gctgcgggat cgacgcccgc ccgcaactgg
ccggcctcac gggcgtcctg caacggcacc 4260tccagcaggt gggtccagtc cacgtagggc
aggggcagct ccgcgtcgat gaaggcgcgc 4320tcactctgca gccgggcacc ggcctgcatc
acggggtcgt cgcggaaggc ctgcgccgcg 4380cgatggagca tctcctcgac cgtctccagc
ggtgtgaagc cctggatgcg gatctcttcc 4440atcgctgcgg gccagcgcgc gtagtgctcc
tccaccacgg cgatggccag tgattccttg 4500ctggggaagt ggaagtacac cgcgcccttg
gtcatgccga cacgctcggc aacgtccttg 4560acggaggccc ctcggtagcc gcgcgacgcg
aagatctccg acgccgcctc caggatctgg 4620gcccgggtct tgatcgaccg ctcctgctgc
gggtgaggtc cacgcggcac gttggaccgg 4680accgcgaaag gcgtgggttg ttgggcgctc
gtcatggctg ccttccttcg tgtgtgcggg 4740ccctgcgggc gccatgctgg tgcgacccgg
gtcggcacgg aaacccattg cataatacct 4800tcccgcaggt atatttctct cggtcagctt
accgatcccg gctgtcttgc agcgcggcaa 4860gccagccggt ggtcccgtac gaggacacac
cgggagatgt agcgccccta tgaaccatac 4920aaaccgtctt ttactgcccg ccccccacga
cctcctgttc gacggctgcc ctccgctctc 4980cttcgcgcgc cccctgccgc cggccgacgt
acacaaggct gccgcggcag aagtactcct 5040caccgacgcg cgaccgctgg gcgagaaccg
gttcgccgtc gccgccctgt ggccccgaaa 5100caccttcctc gcgcaccgcg cgacatcgag
cccgtgcgac cccctcctag ccgcggagac 5160catacgacag tcggcgatcc acctctcaca
caccttctgt gacgtgccca taggccacca 5220cttcgtgctg tcgggcctcg atcttgatct
cgatctccct gtctgggact ccggcccgct 5280gcctgtcgtc ctggacgtca cgagtacgaa
gaccaccacg aacccgcgcc ggatggccag 5340ggcgttgaac gccgacgtgt acgtggccgg
actccaccgc ggccgctgcg ccatccgctt 5400cgaggtcctc gccccccggc ggtacgcgat
gatccgcgat cgcgccaggc gggcagagcg 5460ccccgcacag caggcagccg caggcgcggc
gaccgccctt cctcccgaaa ccgtcggctt 5520ccacgacgac ctgcacgtcc tgcttgccac
cgcgcaaggc ctgcccgata cagcctggca 5580actgcggctg cgccgggacc atccggtcct
cttcgaccac gaatcggacc acatttccgg 5640catggcactg ctggaggcct gccgtcaggc
cgccacagcc ctcaccccac ccgcgcccgg 5700agccttcggc ccgcggcagg tggccctcac
ggccgtcgca agttcctacc aggcattcgg 5760cgaactcgac tcccctgtca ccatcaccac
cctccccgcc gcccacgggc acagcccgga 5820cagcggcaca cgtacgctgc aactcaccgc
ccggcaaggc agccgcacgc tcatcaccgc 5880gaccgtcacc acgacgacga cagccgggac
gggaagcccc gggcccaccg tcccgcacca 5940cggcgaccag acgaaggcgg tggcctcatg
accgcgccgc tcagggagtt ttcccgcgac 6000ggggtggaac acgccgtggc tctcgcgcac
cgggaccacg gagtccagga gacacaccga 6060cgactcacag acgacgtggc ccgggcggtc
accgacgtcg gctttccacg ccacttcgtc 6120ccccggcgct tcggcggccg cgccgggacc
ttcggcgaac tgctcacggc ggcgaccacg 6180ctcgcccgga cctgcgccgc caccgcctgg
tgcgccacgc tgtatgcggc ccatgggcgg 6240ctggcctcgt acctgcccga aaaggcccaa
cgcgaactgt ggcactcctc gcccgacgcc 6300cgtatcgccg cggccatcat gcccccgtcg
ggcgaggcca acctcgagcc gggcggctgg 6360cggctcaccg ggcgctgggg attcgccagc
ggcgtcgacc acgccgactg ggtgctgctc 6420gcaagctgga caccgggccg gaacgttccc
gagcggcacc ggctgttcgc cgtccccagg 6480gacgagctga cggtcaccga cacctggcac
accctcggca tgcgcgggac cggcagcaac 6540accgtcgagg cggacggggt gctggttccc
cgccaccgca cctgcacctt gtcggacctg 6600ctgctccccc tgcccggctc ggcccgctgc
cacacggtgc cctacgccat ggtgggcgcg 6660ctgatgttcg cctttccggt tctcggggcg
gcccgcggag ctcttgacgc ctggacacac 6720gccgccacag aacgacaggg caccgcagtg
cccccggcgt ccaacaccct gaccagagca 6780gcagcacgca tacgcgccgc aggcctgctc
ctggaggcag cggcagagcg ggccgaccac 6840gcgccagtga ccccgctcct cgtcgccgag
gggcagcggg acgccgccgc ggctgtggaa 6900ctgtgctccg aagccgtcga ccaactgctg
cgcgcctcgg gatcacgtgg ccaggcagaa 6960gacgacccgg tacagcgcca ctggcgggac
atcaccaccg ctgccacgca cagggccctg 7020agcatcgacg cagcagccgg cgcatacacg
cctgctctct tcgaccgtgc ggacccatcg 7080accggcgtcg gccccgccga gccgggcggc
ccggccggcc cacccggcag aaccgcgacc 7140gcaccagaca cagaaagacg tacggcgtga
tgcccgagcc cccgcgcgaa cgtcgcacag 7200ccgcgaaccg gtcgcccgcc atacgcccca
tcgccttctt cgatgtggac gagacactga 7260tcaccgccaa gagcatgctc gacttcgcac
ggcaggcgcc gcacagcttg cgggacgaca 7320tcaccgcgca ggcgagcggg caacgccaca
gtgctgatgc cgatctgacg gcgatgcggc 7380gccgaggggc gagccgggtg gagatgaacc
gcgtctacta ccggcgctac gccggtgttt 7440ccctggctcg cctgcaggag gccggccgcg
actggtacca cgcttaccgc acgcggccgg 7500acggctacgt cagggccggc ctggcggcac
tcgctcggca ccgccgggcc ggccacacga 7560tcgttctgat ctcgggctcg gcccggccac
tgctcacgcc cctggcgcag gacctcggcg 7620cggaccggat tctgtgcacc gagcagttcg
ccgacgccca gggcgtcctc accggcgagg 7680tgaatcgccc gatgatcggc gaagcgaagg
ctgaggccgt gaccgaagtg atggcgaaga 7740ggggggtggt gccggcagac tgctttgcct
acggggacca cgagagcgac ttcggcatgc 7800tgcaggccgt ggggaacccg gtcgtggtcg
gcacggacct cgtgctggtg cgccacgccc 7860aggggtccaa ctggccggtg ctgcccgccg
acgcgggacc ccgctgtgca tgcgcacggc 7920gtccaggccc gctaggccac gacgatccgt
cggccatcgg ttgaatcctg ccgcgcggta 7980gccgtgctgc ctccactttt gcgccgatga
ctgggacatc gtccacgtgc gccgaccgcc 8040cccactaaca taccttcccg agggtatgtt
ttccgggccc gttggctcac gaccttgaga 8100ggactcgggc gttggtgaag caggcaaggg
cgatgcgcac ccgggaccag gtgctggatg 8160cggcggctga agagttcgcc ctgcacggct
atgcaggcac caatctggca acggtcgccg 8220tacgcacagg catgacgaag ggcgctctct
acgggcactt cccatccaag aaggcgctcg 8280ccgacgaact ggtgagccag tcgacagaga
catggaacac catcggccgc tccatcgccg 8340agaccgcctg cgcgcccgaa accgccttgc
gtgccctcgt gctggccgta agccgtcaaa 8400tgaagcacga catccggttc cgtgcggcgc
tgcggctggc agcggactgc accatgcccg 8460cgggcggggc gccggatctc ctggaccgca
tccgccgcga gatggccgcg gccgcccgtg 8520acacccagca acagcaagcc ccctactcac
ccttggccac gcagccaccg gacgtggtgg 8580tccacctgct gctgaccgtt gcctatgggt
tgtcgttcgc ggctgaacga ggggcgccgg 8640gacgctcacc tgccacgact gacaaggtgt
gggaactcct gcttactgcg ctgcagttgg 8700aggacatttc aacctgccac aactgactgc
ggcgaccagg atccggcgga caacgagctc 8760cggcccaggc agaccatgcc ggctccgggc
tcacgggcac cggcaaagaa gagcgccaga 8820gcgcgtggcg tccggttacg gatcaggcag
tccgtttcct gcgggggaga gcgacagcga 8880ggtcaggccg gcggcagacg gagcgagcgc
agccgctcgg cgacggcgaa ggagtcgaga 8940cgcagggcgg tgatgtcgga gatcaggtcc
cgccaggcct cgaattttcg tgttcacccg 9000ctccccgtgc atcccgaggt acgccacggc
cacgccgaag ccgtacagct cgttgcgggc 9060gggcaggggc cggagcacga cgcactcctc
cagcatcacg gccgcccgcc agaaggcgtc 9120cgggtgatgc tggtcgtgac ggggcgggtc
gaccctgtgc cgggcggcta ggccgacgaa 9180gacggagtag tcgctgaccg cgagatcgtc
cagcagctcg gcctggcgct ccaggaacca 9240ccggtagtcg acgtagtact cggcaggcac
aggtcaggcc gcccgcgggg cggtctgcgc 9300ctgcgcgggg ccgccgtact ccgcgtcgaa
ggcggcgacg gtctccgggg tgccggtgac 9360ccggcggaag atctccgcgc cctcggccag
cgcgcgctcg cgccgcttct cctcggcgac 9420ctgcgcgagg taggcgtcca gcggcagccc
ggccgcgtcg gccagggccc gcagctcgtc 9480cgcagtggtg tcgtcgacct cgatgccagc
catgtctcga g 952120265PRTStreptomyces coelicolor
20Val Met Pro Glu Pro Pro Arg Glu Arg Arg Thr Ala Ala Asn Arg Ser1
5 10 15Pro Ala Ile Arg Pro Ile
Ala Phe Phe Asp Val Asp Glu Thr Leu Ile 20 25
30Thr Ala Lys Ser Met Leu Asp Phe Ala Arg Gln Ala Pro
His Ser Leu 35 40 45Arg Asp Asp
Ile Thr Ala Gln Ala Ser Gly Gln Arg His Ser Ala Asp 50
55 60Ala Asp Leu Thr Ala Met Arg Arg Arg Gly Ala Ser
Arg Val Glu Met65 70 75
80Asn Arg Val Tyr Tyr Arg Arg Tyr Ala Gly Val Ser Leu Ala Arg Leu
85 90 95Gln Glu Ala Gly Arg Asp
Trp Tyr His Ala Tyr Arg Thr Arg Pro Asp 100
105 110Gly Tyr Val Arg Ala Gly Leu Ala Ala Leu Ala Arg
His Arg Arg Ala 115 120 125Gly His
Thr Ile Val Leu Ile Ser Gly Ser Ala Arg Pro Leu Leu Thr 130
135 140Pro Leu Ala Gln Asp Leu Gly Ala Asp Arg Ile
Leu Cys Thr Glu Gln145 150 155
160Phe Ala Asp Ala Gln Gly Val Leu Thr Gly Glu Val Asn Arg Pro Met
165 170 175Ile Gly Glu Ala
Lys Ala Glu Ala Val Thr Glu Val Met Ala Lys Arg 180
185 190Gly Val Val Pro Ala Asp Cys Phe Ala Tyr Gly
Asp His Glu Ser Asp 195 200 205Phe
Gly Met Leu Gln Ala Val Gly Asn Pro Val Val Val Gly Thr Asp 210
215 220Leu Val Leu Val Arg His Ala Gln Gly Ser
Asn Trp Pro Val Leu Pro225 230 235
240Ala Asp Ala Gly Pro Arg Cys Ala Cys Ala Arg Arg Pro Gly Pro
Leu 245 250 255Gly His Asp
Asp Pro Ser Ala Ile Gly 260
26521400PRTStreptomyces coelicolor 21Met Thr Ala Pro Leu Arg Glu Phe Ser
Arg Asp Gly Val Glu His Ala1 5 10
15Val Ala Leu Ala His Arg Asp His Gly Val Gln Glu Thr His Arg
Arg 20 25 30Leu Thr Asp Asp
Val Ala Arg Ala Val Thr Asp Val Gly Phe Pro Arg 35
40 45His Phe Val Pro Arg Arg Phe Gly Gly Arg Ala Gly
Thr Phe Gly Glu 50 55 60Leu Leu Thr
Ala Ala Thr Thr Leu Ala Arg Thr Cys Ala Ala Thr Ala65 70
75 80Trp Cys Ala Thr Leu Tyr Ala Ala
His Gly Arg Leu Ala Ser Tyr Leu 85 90
95Pro Glu Lys Ala Gln Arg Glu Leu Trp His Ser Ser Pro Asp
Ala Arg 100 105 110Ile Ala Ala
Ala Ile Met Pro Pro Ser Gly Glu Ala Asn Leu Glu Pro 115
120 125Gly Gly Trp Arg Leu Thr Gly Arg Trp Gly Phe
Ala Ser Gly Val Asp 130 135 140His Ala
Asp Trp Val Leu Leu Ala Ser Trp Thr Pro Gly Arg Asn Val145
150 155 160Pro Glu Arg His Arg Leu Phe
Ala Val Pro Arg Asp Glu Leu Thr Val 165
170 175Thr Asp Thr Trp His Thr Leu Gly Met Arg Gly Thr
Gly Ser Asn Thr 180 185 190Val
Glu Ala Asp Gly Val Leu Val Pro Arg His Arg Thr Cys Thr Leu 195
200 205Ser Asp Leu Leu Leu Pro Leu Pro Gly
Ser Ala Arg Cys His Thr Val 210 215
220Pro Tyr Ala Met Val Gly Ala Leu Met Phe Ala Phe Pro Val Leu Gly225
230 235 240Ala Ala Arg Gly
Ala Leu Asp Ala Trp Thr His Ala Ala Thr Glu Arg 245
250 255Gln Gly Thr Ala Val Pro Pro Ala Ser Asn
Thr Leu Thr Arg Ala Ala 260 265
270Ala Arg Ile Arg Ala Ala Gly Leu Leu Leu Glu Ala Ala Ala Glu Arg
275 280 285Ala Asp His Ala Pro Val Thr
Pro Leu Leu Val Ala Glu Gly Gln Arg 290 295
300Asp Ala Ala Ala Ala Val Glu Leu Cys Ser Glu Ala Val Asp Gln
Leu305 310 315 320Leu Arg
Ala Ser Gly Ser Arg Gly Gln Ala Glu Asp Asp Pro Val Gln
325 330 335Arg His Trp Arg Asp Ile Thr
Thr Ala Ala Thr His Arg Ala Leu Ser 340 345
350Ile Asp Ala Ala Ala Gly Ala Tyr Thr Pro Ala Leu Phe Asp
Arg Ala 355 360 365Asp Pro Ser Thr
Gly Val Gly Pro Ala Glu Pro Gly Gly Pro Ala Gly 370
375 380Pro Pro Gly Arg Thr Ala Thr Ala Pro Asp Thr Glu
Arg Arg Thr Ala385 390 395
40022241PRTStreptomyces coelicolor 22Val Ile Pro Val Arg Leu Pro Pro Pro
Glu Asp Val Ser Ala Glu Thr1 5 10
15Ala Asp Gly Ser Gly Met Ser Met Thr Leu Val Val Ala Ser Leu
Asp 20 25 30His Glu Leu Gly
Pro Met Leu Arg Arg Pro Phe Leu Phe Tyr Gly His 35
40 45Ser Met Gly Ala Leu Val Ala Tyr His Leu Thr Arg
Leu Arg Gln Ser 50 55 60Arg Gly Arg
Pro Leu Pro Glu Arg Leu Leu Ile Gly Ala Tyr Pro Ala65 70
75 80Pro His Leu Pro His Arg Leu Ala
His Cys Thr His Leu Pro Asp Glu 85 90
95Asp Leu Leu Ala Leu Leu Pro Pro His Pro Ala Gly His Ser
Arg Leu 100 105 110Leu Arg Gln
Ala Pro Gly Leu Ala Thr Ala Thr Ala Ala Arg Leu Arg 115
120 125Leu His Leu Gly Leu Cys Asp Ser Ala Ala Pro
Ala Ala Pro Asn Pro 130 135 140Ala Gln
His Thr Gly His Gly Ser Pro Gln Gly Arg Ser Glu Pro Leu145
150 155 160Arg Cys Pro Val Asp Val Phe
Thr Gly Ile Ser Asp Pro Leu Val Thr 165
170 175Asp Ala Glu Ala Ala Ala Trp Arg His His Thr Arg
Ala Gly Cys Arg 180 185 190Ile
His Arg Ile Pro Gly Gly His Phe Phe Thr Arg Glu Thr Pro Glu 195
200 205Ser Arg Ala Ala Phe Phe Asp Arg Leu
Cys Thr Val Leu Ala Gly Pro 210 215
220Ser Glu Trp Ala Ala Gly Ala Ser Gly Pro Leu Pro Val Thr Val Ala225
230 235
240Ser23373PRTStreptomyces coelicolor 23Met Tyr Pro Glu Thr Leu Gly Phe
Gly Ala Phe Leu Ser Pro Met His1 5 10
15Pro Leu Gly Glu Asn Pro Thr Leu Gln Phe Gln Arg Asp Leu
Glu Leu 20 25 30Ile Glu Leu
Leu Asp Arg Leu Asp Tyr Asn Glu Phe Trp Val Gly Glu 35
40 45His His Ser Met Gly Trp Asn Thr Ile Gly Ser
Pro Glu Leu Met Val 50 55 60Ala Ala
Ala Ala Glu Arg Thr Arg Arg Ile Thr Leu Ala Thr Gly Val65
70 75 80Met Thr Leu Pro Tyr His His
Pro Phe Met Val Ala Ser Arg Ala Val 85 90
95His Leu Asp His Leu Thr Arg Gly Arg Phe Val Leu Gly
Val Gly Ala 100 105 110Gly Gly
Ile Pro Thr Asp Ala Arg Met Ile Gly Arg Glu Met Ser Glu 115
120 125Leu Arg Thr Met Phe Gly Glu Ala Leu Glu
Ala Val Val Ala Leu Val 130 135 140Asn
Gly Glu Glu Arg Val Thr Lys Lys Thr Ser Trp Phe Thr Leu Lys145
150 155 160Asp Ala Lys Leu Gln Leu
Ser Pro Tyr Arg Ala Ser Gly Leu Glu Ile 165
170 175Ala Ala Ala Ser Val Ala Ser Gly Asn Ser Met Arg
Leu Ala Gly Arg 180 185 190Tyr
Gly Ile Ser Thr Val Ser Phe Gly Ala Pro Arg Pro Gly His Pro 195
200 205Arg Pro Asp Met Arg Thr Gln Trp Ser
Tyr Ala Glu Glu Ala Ala Ala 210 215
220Glu Gln Gly Thr Thr Val Asp Arg Arg Asn Trp Arg Ile Thr Leu Pro225
230 235 240Val Tyr Val Ala
Glu Thr Arg Glu Gln Ala Leu Ala Asp Val Arg Glu 245
250 255Gly Tyr Asp Arg Trp Ala Tyr Gly Tyr Trp
Gly Asp Ile Arg Gly Leu 260 265
270Asp Val Ser Val Pro Gly Val Lys Arg Ala Gln Ala Leu Glu Ala Ala
275 280 285Val Asp Ala Gly Ser Ala Ile
Val Gly Ser Val Glu Asp Val Val Ala 290 295
300Gly Val Glu Arg Leu Arg Glu Glu Val Gly Gly Phe Gly Thr Leu
Leu305 310 315 320Val Tyr
Ala Gln Asp Trp Ala Asp Trp Glu Lys Thr Lys Arg Ser Tyr
325 330 335Asp Leu Leu Ala Arg Tyr Val
Ala Pro His Phe Thr Gly Ser Thr Arg 340 345
350Arg Leu Tyr Glu Ser Val Gln Trp Tyr Gln Asp Asn Arg Asp
Leu Phe 355 360 365Pro Gln Leu Ile
Pro 37024393PRTStreptomyces coelicolor 24Met Ala Thr Glu Pro Ile Arg
Ile Gly Val Val Gly Ala Ser Pro Asp1 5 10
15Arg Gly Trp Ala Ala Asp Ala His Leu Pro Ala Leu Gln
His Leu Pro 20 25 30Gln Tyr
Lys Ile Thr Ala Val Gly Thr Arg Arg Ala Asp Ser Ala His 35
40 45Arg Ala Ala Arg Arg Tyr Gly Ala Thr His
Ala Phe Thr Asp Pro Arg 50 55 60Ser
Leu Ala Ala His Pro Asp Val Glu Leu Val Ala Ile Val Val Lys65
70 75 80Val Pro Asp His Ala Arg
Leu Val Glu Ala Ala Leu Ala Ala Gly Lys 85
90 95His Val Leu Cys Glu Trp Pro Leu Ala Arg Thr Thr
Glu Glu Ala Ala 100 105 110Gln
Leu Thr Ala Ala Ala His Gly Ala Gly Val Val Asn Ala Val Gly 115
120 125Leu Gln Ala Arg His Thr Pro Thr Val
Val Arg Ala Arg Glu Leu Ile 130 135
140Arg Gln Gly Tyr Val Gly Arg Val Thr Ser Val Thr Val Tyr Ser Thr145
150 155 160Arg Gly Val Ala
Ala Gly Gly Arg Leu Pro Ala Ala Phe Ala Tyr Thr 165
170 175Leu Asp Ser Thr Asn Gly Ala Gly Thr Phe
Glu Val Ala Gly Gly His 180 185
190Thr Leu Asp Ala Val Gln Tyr Leu Leu Gly Arg Glu Met Thr Gly Leu
195 200 205Ser Ala Ala Leu Ser Val Gln
His Pro Arg Ile Thr Leu Asp Glu Asp 210 215
220Ala Arg Gln Thr Gly Ala Thr Ser Pro Asp His Val Ala Leu His
Ala225 230 235 240Thr Leu
Glu Gly Gly Ala Ala Leu Val Val His Ile His Asp Ala Lys
245 250 255Asn Ser Gly Ala Gly Thr Arg
Ile Glu Ile Ser Gly Thr Gln Gly Glu 260 265
270Leu Ala Ile Val Ser Thr Gly Pro Arg Ser Gly Ser Gly Leu
Gln Ile 275 280 285Ser Glu Leu Ala
Leu Leu Gly Ala Gln Gly Thr Glu Pro Ser Gly Gln 290
295 300Glu Leu Pro Phe Pro Gly Ser Trp Gly Thr Ala Val
Pro Ala Asp Gly305 310 315
320Leu Asp Ala Ala Gln His Thr Met Ala Val Gln Tyr Ala Ala Leu Ala
325 330 335Ala Asp Ile Arg Glu
Gly Gly Ser Arg Val Pro Arg Phe Ala Asp Gly 340
345 350Ile Glu Leu His Arg Leu Leu Asp Ala Val Arg Leu
Ser Ser Ala Thr 355 360 365Gly Cys
Arg Leu Glu Arg Arg Ala Gly Glu Arg Trp Pro Val Ser Ser 370
375 380Pro Trp Pro Arg Arg Arg Arg Ser Arg385
3902538DNAArtificialPrimer 25tttttctaga tgattaggag gaccgtgcgc
ggcgcgat 382639DNAArtificialPrimer
26ttttaagctt atcgcggtca tggacgacgt ggtacgtgt
392720DNAArtificialOligonucleotide linker 27catggaggaa gcttatgatc
202820DNAArtificialOligonucleotide linker 28gatcataagc ttcctccatg
202937DNAArtificialPrimer
29tataagcttg gtgaactcct tcggcgagtg gttcgga
373036DNAArtificialPrimer 30tatggtaccg gggagaactc cttgggatac ttcctg
363136DNAArtificialOligonucleotide primer
31attactagtt cgccgagcgg ctgcgctcgc tccgtc
363227DNAArtificialOligonucleotide primer 32ccgccgacgc gggaccccgc tgtgcat
273333DNAArtificialOligonucleotide primer 33aatcactggc catcgccgtg
gtggaggagc act
333436DNAArtificialOligonucleotide primer 34tttcatatgc gcccgcgctc
ccagtctctt ctgcca
363536DNAArtificialOligonucleotide primer 35tataagcttg gggagaactc
cttgggatac ttcctg 36
User Contributions:
comments("1"); ?> comment_form("1"); ?>Inventors list |
Agents list |
Assignees list |
List by place |
Classification tree browser |
Top 100 Inventors |
Top 100 Agents |
Top 100 Assignees |
Usenet FAQ Index |
Documents |
Other FAQs |
User Contributions:
Comment about this patent or add new information about this topic:
| People who visited this patent also read: | |
| Patent application number | Title |
|---|---|
| 20120225269 | RADIO WAVE ABSORBING COATING |
| 20120225268 | INSULATIVE ELEMENTS AND METHODS OF FORMING THE SAME |
| 20120225267 | WEB SUBSTRATES HAVING WIDE COLOR GAMUT INDICIA PRINTED THEREON |
| 20120225266 | WEB SUBSTRATES HAVING WIDE COLOR GAMUT INDICIA PRINTED THEREON |
| 20120225265 | MULTIVARIATE COLOR SYSTEM WITH TEXTURE APPLICATION |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 | 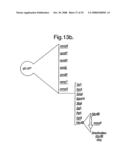 |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 | 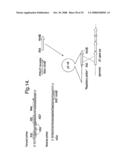 |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |
| Similar patent applications: | |
| Date | Title |
|---|---|
| 2010-07-22 | Methods and apparatuses relating to cell culture media |
| 2009-05-28 | Splicing-mediated regulation of gene expression |
| 2009-06-11 | Methods for analysis of gene expression |
| 2009-06-18 | Assessment of asthma and allergen-dependent gene expression |
| 2010-01-21 | Composition and method for determination of ck19 expression |
| New patent applications in this class: | |
| Date | Title |
|---|---|
| 2022-05-05 | Engineered cd47 extracellular domain for bioconjugation |
| 2019-05-16 | High cell density anaerobic fermentation for protein expression |
| 2019-05-16 | Polynucleotide encoding fusion of anchoring motif and dehalogenase, host cell including the polynucleotide, and use thereof |
| 2019-05-16 | Cell culture method, medium, and medium kit |
| 2018-01-25 | Protein expression strains |
| Top Inventors for class "Chemistry: molecular biology and microbiology" | |
| Rank | Inventor's name |
|---|---|
| 1 | Marshall Medoff |
| 2 | Anthony P. Burgard |
| 3 | Mark J. Burk |
| 4 | Robin E. Osterhout |
| 5 | Rangarajan Sampath |