Patent application title: Compositions and Methods Relating to STOP-1
Inventors:
Heidi Ackerly (Belmont, CA, US)
Avi Ashkenazi (San Mateo, CA, US)
Avi Ashkenazi (San Mateo, CA, US)
David Eberhard (San Francisco, CA, US)
Gretchen Frantz (San Francisco, CA, US)
Dorothy French (San Carlos, CA, US)
Dorothy French (San Carlos, CA, US)
Germaine G. Fuh (Pacifica, CA, US)
Jo-Anne S. Hongo (Redwood City, CA, US)
Jo-Anne S. Hongo (Redwood City, CA, US)
Chingwei V. Lee (Foster City, CA, US)
Chingwei V. Lee (Foster City, CA, US)
Scot A. Marsters (San Carlos, CA, US)
Helga A. Raab (San Francisco, CA, US)
Evgeny Varfolomeev (San Bruno, CA, US)
Beni B. Wolf (Redwood City, CA, US)
Robert Maurice Pitti (El Cerrito, CA, US)
Liliana Soroceanu (San Francisco, CA, US)
Assignees:
Genentech, Inc.
IPC8 Class: AC07K1618FI
USPC Class:
5303873
Class name: Globulins immunoglobulin, antibody, or fragment thereof, other than immunoglobulin antibody, or fragment thereof that is conjugated or adsorbed chimeric, mutated, or recombined hybrid (e.g., bifunctional, bispecific, rodent-human chimeric, single chain, rfv, immunoglobulin fusion protein, etc.)
Publication date: 2011-05-12
Patent application number: 20110112279
Inventors list |
Agents list |
Assignees list |
List by place |
Classification tree browser |
Top 100 Inventors |
Top 100 Agents |
Top 100 Assignees |
Usenet FAQ Index |
Documents |
Other FAQs |
Patent application title: Compositions and Methods Relating to STOP-1
Inventors:
Scot A. Marsters
Liliana Soroceanu
Gretchen Frantz
Jo-Anne S. Hongo
Beni B. Wolf
Avi Ashkenazi
Chingwei V. Lee
Dorothy French
HEIDI ACKERLY
David Eberhard
Germaine G. Fuh
Helga A. Raab
Evgeny Varfolomeev
Robert Maurice Pitti
Agents:
Assignees:
Origin: ,
IPC8 Class: AC07K1618FI
USPC Class:
Publication date: 05/12/2011
Patent application number: 20110112279
Abstract:
The present invention provides novel polypeptides, antibodies,
antagonists, agonists, potentiators, nucleic acid molecules, compositions
and methods relating to the STOP-1 polypeptide that are useful for
treating and preventing diseases and for medical diagnosis and research.
The present invention also provides consensus sequences and specific
sequences for antibodies that specifically bind to STOP-1 that are useful
in the methods described herein.Claims:
1. A monoclonal antibody that specifically binds to amino acids 94-243 of
human STOP-1.
2. The monoclonal antibody of claim 1, wherein the antibody potentiates STOP-1 binding to cells.
3. The monoclonal antibody of claim 1, wherein the monoclonal antibody binds to an oligomeric form of human STOP-1.
4. The monoclonal antibody of claim 1 selected from the group consisting of: (i) a monoclonal antibody comprising (a) a VH-CDR1 comprising the amino acid sequence TISGSD (SEQ ID NO:8); (b) a VH-CDR2 comprising the amino acid sequence GRISPYGGNTN (SEQ ID NO:9); and (c) a VH-CDR3 comprising the amino acid sequence CARVGGLKLLFDY (SEQ ID NO:10); (ii) a monoclonal antibody comprising (a) a VH-CDR1 comprising the amino acid sequence TITNSD (SEQ ID NO:11); (b) a VH-CDR2 comprising the amino acid sequence ATIYPYGGYTY (SEQ ID NO:12); and (c) a VH-CDR3 comprising the amino acid sequence CARGGGMDGYVMDY (SEQ ID NO:13); (iii) a monoclonal antibody comprising (a) a VH-CDR1 comprising the amino acid sequence TISGSW (SEQ ID NO:17); (b) a VH-CDR2 comprising the amino acid sequence AWIAPYSGATD (SEQ ID NO:18); and (c) a VH-CDR3 comprising the amino acid sequence CAREGGLYWVFDY (SEQ ID NO:19); and (iv) a monoclonal antibody comprising (a) a VH-CDR1 comprising the amino acid sequence TISNYG (SEQ ID NO:20); (b) a VH-CDR2 comprising the amino acid sequence GRISPSNGSTY (SEQ ID NO:21); and (c) a VH-CDR3 comprising the amino acid sequence CAKCSVRFAY (SEQ ID NO:22).
5. The monoclonal antibody of claim 4 comprising (a) a VH-CDR1 comprising the amino acid sequence TISGSD (SEQ ID NO:8); (b) a VH-CDR2 comprising the amino acid sequence GRISPYGGNTN (SEQ ID NO:9); and (c) a VH-CDR3 comprising the amino acid sequence CARVGGLKLLFDY (SEQ ID NO:10).
6. The monoclonal antibody of claim 4 comprising (a) a VH-CDR1 comprising the amino acid sequence TITNSD (SEQ ID NO:11); (b) a VH-CDR2 comprising the amino acid sequence ATIYPYGGYTY (SEQ ID NO:12); and (c) a VH-CDR3 comprising the amino acid sequence CARGGGMDGYVMDY (SEQ ID NO:13).
7. The monoclonal antibody of claim 4 comprising (a) a VH-CDR1 comprising the amino acid sequence TISGSW (SEQ ID NO:17); (b) a VH-CDR2 comprising the amino acid sequence AWIAPYSGATD (SEQ ID NO:18); and (c) a VH-CDR3 comprising the amino acid sequence CAREGGLYWVFDY (SEQ ID NO:19).
8. The monoclonal antibody of claim 4 comprising (a) a VH-CDR1 comprising the amino acid sequence TISNYG (SEQ ID NO:20); (b) a VH-CDR2 comprising the amino acid sequence GRISPSNGSTY (SEQ ID NO:21); and (c) a VH-CDR3 comprising the amino acid sequence CAKCSVRFAY (SEQ ID NO:22).
9. The monoclonal antibody of claim 4 comprising the amino acid sequence of: (a) the heavy chain of FIG. 27 (amino acids 21-250 of SEQ ID NO:93); (b) the heavy chain of FIG. 28 (amino acids 21-247 of SEQ ID NO:96); (c) the heavy chain of FIG. 29 (amino acids 21-250 of SEQ ID NO:99); (d) the heavy chain of FIG. 30 (amino acids 21-251 of SEQ ID NO:102); or (e) the heavy chain of FIG. 34 (amino acids 17-468 of SEQ ID NO:112).
10. The monoclonal antibody of claim 9, further comprising the amino acid sequence of: (a) the light chain of FIG. 27 (amino acids 24-239 of SEQ ID NO:92); or (b) the light chain of FIG. 33 (amino acids 20-233 of SEQ ID NO:110).
11. The monoclonal antibody of claim 4, wherein the antibody comprises the light and heavy chain sequences of an antibody selected from the group consisting of S7 encoded by the nucleic acid molecule deposited with ATCC as designation V0350-4-S7, S4 encoded by the nucleic acid molecule deposited with ATCC as designation V0350-2b-S4, S9 encoded by the nucleic acid molecule deposited with ATCC as designation V0350-2b-S9, and S16 encoded by the nucleic acid molecule deposited with ATCC as designation V0350-4-S16.
12. A monoclonal antibody that potentiates STOP-1 binding to cells.
13. A monoclonal antibody that specifically binds to STOP-1, wherein the binding of the antibody to STOP-1 can be inhibited by a second monoclonal antibody selected from the group consisting of S7 encoded by the nucleic acid molecule deposited with ATCC as designation V0350-4-S7, S4 encoded by the nucleic acid molecule deposited with ATCC as designation V0350-2b-S4, S9 encoded by the nucleic acid molecule deposited with ATCC as designation V0350-2b-S9, and S16 encoded by the nucleic acid molecule deposited with ATCC as designation V0350-4-S16.
14. The antibody according to any one of claims 1-8 and 12-13, wherein the antibody is a chimeric antibody, humanized antibody, antibody fragment, or bispecific antibody.
Description:
[0001] This application is a divisional application of U.S. application
Ser. No. 10/553,491, filed Apr. 18, 2007, now U.S. Pat. No. 7,799,899,
which is the National Stage of International Application No.
PCT/US2004/011793, filed Apr. 16, 2004, which claims the benefit of U.S.
Provisional Application No. 60/463,656, filed Apr. 16, 2003. The
foregoing applications are incorporated by reference in their entirety.
FIELD OF THE INVENTION
[0002] The present invention is directed to STOP-1 polypeptides, antibodies, nucleic acid molecules, antagonists, agonists, potentiators and compositions relating to STOP-1, and methods of making and using the same, including methods for diagnosing and treating of tumors in mammals. The present invention further relates to the diagnosis and treatment of disorders involving angiogenesis and vasculogenesis (e.g., cardiovascular as well as oncological disorders).
BACKGROUND AND INTRODUCTION OF THE INVENTION
[0003] Uncontrolled cell growth is the cause of many illnesses in a variety of cell types. For example, cancer occurs when there is an increase in the number of abnormal, or neoplastic, cells derived from a normal tissue that proliferate to form a tumor mass. The tumor cells often invade the adjacent tissues and can spread via the blood or lymphatic system to regional lymph nodes and to distant sites via a process called metastasis. In a cancerous growth, a cell proliferates under conditions in which normal cells would not grow. Cancer manifests itself in a wide variety of forms, characterized by different degrees of invasiveness and aggressiveness. Malignant tumors (cancers) are the second leading cause of death in the United States, after heart disease (Boring et al., CA Cancel J. Clin. 43:7 (1993)).
[0004] Much research has been devoted to discovering new treatments for cell proliferative disorders, such as cancer. Despite recent advances, there is a great need to identify and understand the role of new cellular targets for modulating cell proliferation and to develop alternative or more effective methods of treatment and therapeutic and diagnostic agents.
[0005] There is also a need to develop alternative therapeutics and methods for treating specific cell types and for treating illnesses caused by or associated with abnormal cell proliferation, such as cancers. For example, desmoplasia is the hyperplasia of fibroblasts and disproportionate formation of fibrous connective tissue, especially in the stroma of carcinomas. Desmoplasia is a hallmark of tumor invasion and malignancy. Desmoid tumors and abdominal fibroids are nodules or relatively large masses of unusually firm scarlike connective tissue resulting from active proliferation of fibroblasts, occurring most frequently in the abdominal muscles of women who have borne children; the fibroblasts infiltrate surrounding muscle and fascia.
[0006] In post-natal life, vasculogenesis (endothelial cells forming a primary tubular network) and angiogenesis (the growth or sprouting of new blood vessels from existing vessels) play critical roles in the pathophysiology of neoplastic disorder (Semenza, G. L., (2003) Ann. Rev. Med. 54:17-28). The distinction between vasculogenesis and angiogenesis is not absolute and they overlap (Ribatti, D et al., (2001) Mech. Dev. 100:157-163). Both require endothelial cell proliferation, migration, three-dimensional reorganization of newly formed aggregates and use similar extracellular matrix adhesive mechanisms (Ribatti, supra). Use of anti-angiogenic therapies such as the antibody against vascular endothelial growth (VEGF) called Avastin have been shown to be useful in treating cancers.
[0007] Another cellular protein, referred to herein as STOP-1 or UNQ762, has been shown to be overexpressed in certain tumors (e.g., WO 01/163318, WO 01/68848, WO 02/00690, WO 02/08284, WO 02/16602, WO 02/42487). Polyclonal antibodies against STOP-1 have been reported (e.g., WO 02/42487). Although there has been some discussion of targeting STOP-1 to treat cancers and diseases associated with angiogenesis (e.g., WO 01/163318, WO 01/68848, WO 02/00690, WO 02/08284, WO 02/16602, WO 02/42487, WO 00/71581, WO 02/00690), there is a need to further explore the biology of the STOP-1 protein to identify alternative and more effective therapeutic agents and methods for diagnosis and treatment of uncontrolled cell growth and diseases caused by, associated with or complicated by excessive and insufficient angiogenesis.
[0008] The present invention addresses these needs and others by providing new STOP-1 polypeptides, antibodies, nucleic acid molecules, compositions and methods that incorporate further knowledge about the STOP-1 protein. Among other things, the present disclosure shows that STOP-1 is overexpressed in the stroma of several tumor types. The present disclosure shows that overexpression of STOP-1 alone can be tumorigenic. Further, the present disclosure demonstrates that the STOP-1 protein can be secreted and that secretion is required for tumorigenesis. Still further, the present disclosure shows that the glycosylation state of STOP-1 affects whether it is secreted and that elimination of a N-glycosylation site, e.g., by substituting the amino acid at position 186 (Asn) with alanine results in loss of secretion. The present disclosure shows that disulfide bonding between STOP-1 proteins can occur at a cysteine 55 in culture in the triple helix domain of STOP-1. Additionally, the present disclosure shows that the STOP-1 protein can form a complex with itself as a dimer, trimer and hexamer and that the c-terminus of the protein is sufficient for oligomerization, whereas a region related to the triple helix domain of collagen is not required. The present disclosure also shows a plurality of agents that specifically bind to STOP-1, including the C-terminal region and N-terminal region of the protein as well as nucleic acid and protein sequences encoding them. Further, the present disclosure shows that STOP-1 expression can be modulated by overexpression of proteins in the WNT signalling pathway that are know to cause breast cancer in mice, e.g., the overexpression of WNT. Additionally, the present disclosure shows that STOP-1 can be cleaved by proteases that are overexpressed in the same tumors as STOP-1, e.g., MMP-9. Further, the present disclosure shows that a method for producing STOP-1 polypeptides by expressing the polypeptides in proteoglycan synthesis deficient cell lines. The present invention shows that STOP-1 binds to the surface of cells, such as cancer cells and endothelial cells. The present invention provides antagonistic molecules that can inhibit the interaction of STOP-1 with the surface of cells. The present invention provides molecules that can potentiate the binding of STOP-1 with the surface of cells. The present invention also relates to the role of STOP-1 in angiogenesis and vasculogenesis and methods and compositions for treating disorders for which treatment would be improved by modulating angiogenesis and vasculogenesis. This data and others provided herein, together with other disclosure of in present application, teach new, better and/or alternative methods for using the STOP-1 protein or compositions relating thereto.
SUMMARY OF THE INVENTION
[0009] The present invention provides new therapeutic agents, diagnostic agents and methods for treating or preventing uncontrolled cell proliferation, including cancer, and other diseases by targeting the activity, expression and regulation of STOP-1. The present invention provides new therapeutic agents, diagnostic agents and methods for treating a any medical condition having suboptimal, excessive or inappropriate angiogenic or vasculogenic events by targeting the activity, expression and regulation of STOP-1.
[0010] According to one embodiment, the present invention provides a monoclonal antibody that specifically binds to an oligomeric form of human STOP-1. According to another embodiment, the present invention provides, a monoclonal antibody that specifically binds to amino acids 33-53 or 33-52 of human STOP-1. In yet another embodiment, the present invention includes a monoclonal antibody that specifically binds to amino acids 94-243 of human STOP-1. According to further embodiment, the monoclonal antibody that specifically binds to residues 94-243 of human STOP-1 or residues 33-53 or 33-52 of human STOP-1 also recognizes an oligomeric form of human STOP-1, such as the trimeric form. An antibody according to this invention can be isolated. It is understood that an aforementioned antibody that specifically binds a residue within residues 33-52 or 33-53 of human STOP-1 may also bind to other residues within STOP-1 or non-human equivalents thereof.
[0011] In yet another embodiment, the present invention provides monoclonal antibodies having the biological characteristics of an antibody selected from the group consisting of S7 encoded by the nucleic acid molecule deposited on Mar. 25, 2003 as designation V0350-4-S7, S4 encoded by the nucleic acid molecule deposited on Mar. 25, 2003 as designation V0350-2b-S4, S9 encoded by the nucleic acid molecule deposited on Mar. 25, 2003 as designation V0350-2b-S9, S16 encoded by the nucleic acid molecule deposited on Mar. 25, 2003 as designation V0350-4-S16, F5 encoded by the nucleic acid molecule deposited on Mar. 25, 2003 as designation V0350-5 and 6B12 produced by the hybridoma cell line deposited on Mar. 28, 2003 as designation 6B12.1.7 in the American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, Va. 20110-2209, USA, including the deposited antibodies, antibodies comprising a portion of those antibodies and variants thereof. In another embodiment, the present invention provides antibodies that specifically bind to STOP-1, wherein the binding of the antibodies to STOP-1 can be inhibited (e.g., as observed in a competitive ELISA assay) by a second monoclonal antibody selected from one of the aforementioned deposited antibodies.
[0012] The present invention also relates to antibodies having the following sequences:
[0013] A monoclonal antibody comprising: [0014] (a) a first amino acid sequence comprising:
TABLE-US-00001 [0014] T-I-X1-X2-X3-X4
[0015] wherein X1 is S, N or T; [0016] wherein X2 is G, N, S or A; [0017] wherein X3 is Y, S or T; and [0018] wherein X4 is D or W. [0019] (b) a second amino acid sequence comprising:
TABLE-US-00002 [0019] X1-X2-I-X3-P-X4-X5-G-X6-T-X7 (SEQ ID NO: 115)
[0020] wherein X1 is G or A; [0021] wherein [0022] (1) X2 is an amino acid selected from the group consisting of S, T, A, and X3 is an amino acid selected from the group consisting of R, W and Y; or [0023] (2) X3 is an amino acid selected from the group consisting of S, T, A, and X2 is an amino acid selected from the group consisting of R, W and Y; [0024] wherein X4 is Y or F; [0025] wherein X5 is G, S, T or A; [0026] wherein X6 is N, Y or A; and [0027] wherein X7 is N, Y or D; and [0028] (c) a third amino acid sequence comprising the sequence:
TABLE-US-00003 [0028] (SEQ ID NO: 116) C-X1-X2-X3-G-G-X4-X5-X6-X7-X8-X9-X10-X11
[0029] wherein X1 is A, S or T; [0030] wherein X2 is basic amino acid; [0031] wherein X3 is any amino acid; [0032] wherein X4 is a hydrophobic amino acid; [0033] wherein any one of X5-X8 can be any amino acid or can be missing, and at least one of X5-X8 is an aromatic amino acid or a hydrophobic amino acid; [0034] wherein X9 is an aromatic or hydrophobic amino acid; [0035] wherein X10 is D or A; and [0036] wherein X11 is Y or V.
[0037] According to one embodiment, the monoclonal antibody comprises the light chain sequence of FIG. 34. According to another embodiment, the monoclonal antibody is a full-length IgG.
[0038] According to one embodiment of this invention, the X1 of the first amino acid sequence is S. According to another embodiment of this invention, the X2 the first amino acid sequence is G. According to yet another embodiment of this invention, X3 of the first amino acid sequence is S. According to one embodiment, the first amino acid sequence is a sequence selected from the group consisting of TISGSD (SEQ ID NO:8), TITNSD (SEQ ID NO:11) and TISGSW (SEQ ID NO:17).
[0039] According to yet another embodiment of this invention, X3 of SEQ ID NO:115 is S or A. According to yet another embodiment of this invention, X4 of SEQ ID NO:115 is Y. According to yet another embodiment of this invention, X5 of SEQ ID NO:115 is G or A. According to yet another embodiment of this invention, X6 of SEQ ID NO:115 is N or A. According to one embodiment, SEQ ID NO:115 is a sequence selected from the group consisting of GRISPYGGNTN (SEQ ID NO:9), ATIYPYGGYTY (SEQ ID NO:12) and AWIAPYSGATD (SEQ ID NO:18).
[0040] According to one embodiment of this invention, the X1 of SEQ ID NO:116 is A. According to another embodiment of this invention, the X2 of SEQ ID NO:116 is R. According to yet another embodiment of this invention, X4 of SEQ ID NO:116 is L or M. According to one preferred embodiment of this invention, the aromatic amino acid present in X5-X8 is a tryptophan residue. According to another embodiment, one amino acid of X5-X8 is missing. According to yet another embodiment of this invention, X9 of SEQ ID NO:116 is F. According to one embodiment of this invention, X10 of SEQ ID NO:116 is D. According to one embodiment of this invention, X11 of SEQ ID NO:116 is Y.
[0041] According to one embodiment, the SEQ ID NO:116 is a sequence selected from the group consisting of CARVGGLKLLFDY (SEQ ID NO:10), CARGGGMDGYVMDY (SEQ ID NO:13) and CAREGGLYWVFDY (SEQ ID NO:19).
[0042] An antibody according to this invention can comprise (a) a first amino acid sequence comprising the sequence TISGSD (SEQ ID NO:8); (b) a second amino acid sequence comprising the sequence GRISPYGGNTN (SEQ ID NO:9); and (c) a third amino acid sequence comprising the sequence CARVGGLKLLFDY (SEQ ID NO:10), or a variant of said antibody. Alternatively, an antibody according to this invention can comprise (a) a first amino acid sequence comprising the sequence TITNSD (SEQ ID NO:11); (b) a second amino acid sequence comprising the sequence ATIYPYGGYTY (SEQ ID NO:12); and (c) a third amino acid sequence comprising the sequence CARGGGMDGYVMDY (SEQ ID NO:13); or a variant of said antibody. Alternatively, an antibody according to this invention can comprise (a) a first amino acid sequence comprising the sequence TISGSW (SEQ ID NO:17); (b) a second amino acid sequence comprising the sequence AWIAPYSGATD (SEQ ID NO:18); and (c) a third amino acid sequence comprising the sequence CAREGGLYWVFDY (SEQ ID NO:19); or a variant of said antibody. Alternatively, an antibody according to this invention can comprises (a) a first amino acid sequence comprising the sequence TISNYG (SEQ ID NO:20); (b) a a second amino acid sequence comprising the sequence GRISPSNGSTY (SEQ ID NO:21); and (c) a third amino acid sequence comprising the sequence CAKCSVRFAY (SEQ ID NO:22); or a variant of said antibody. Alternatively, an antibody according to this invention can comprise (a) a first amino acid sequence comprising the sequence TINNYD (SEQ ID NO:14); (b) a second amino acid sequence comprising the sequence GYISPPSGATY (SEQ ID NO:15); and (c) third amino acid sequence comprising the sequence CARMVGMRRGVMDY (SEQ ID NO:16); or a variant of said antibody.
[0043] In a further embodiment, the first, second and third amino acid sequences described above are located in a human heavy chain wherein the first amino acid sequence is at residues 28-33 of the heavy chain according to the Kabat numbering system, the second amino acid sequence is at residues 49-58 of the heavy chain according to the Kabat numbering system and the third amino acid sequence is at residues 92-102 according to the Kabat numbering system.
[0044] In another embodiment, the present invention provides a monoclonal antibody comprising the amino acid sequence of: (a) the heavy chain sequence of FIG. 27; (b) the heavy chain sequence of FIG. 28; (c) the heavy chain sequence of FIG. 29; (d) the heavy chain sequence of FIG. 30; (e) the heavy chain sequence of FIG. 31; or (f) the heavy chain sequence of FIG. 34; or variants thereof. In a further embodiment, the antibodies of this invention further comprise (a) the light chain sequence of FIG. 27, (b) the light chain sequence of FIG. 34; or variants thereof.
[0045] In a further embodiment, the antibodies of this invention are chimeric or humanized antibodies. In another embodiment, the antibodies of this invention are antibody fragments. In yet another embodiment of this invention, the antibodies are conjugated to an agent selected from the group consisting of a stromal targeting agent, a growth inhibitory agent, a cytotoxic agent, a detection agent, an agent that improves the bioavailability and an agent that improves the half-life of the antibody. In another embodiment, the antibody of this invention is a multi-specific antibody having a binding specificity for a STOP-1 polypeptide and one or more binding specificities for any other antigen. According to one embodiment, the other antigen is a cell-surface protein or receptor or receptor subunit. According to one preferred embodiment, the cell-surface protein is a natural killer (NK) receptor receptor. According to a more preferred embodiment, the binding of the antibody to the NK receptor activates the natural killer cell.
[0046] The present invention provides variants and modifications of STOP-1 polypeptide variants. In one embodiment, the STOP-1 polypeptide variant that cannot be secreted from a cell. In another embodiment, said variant is a human STOP-1 polypeptide that is not glycosylated. In a further embodiment, the variant is a human STOP-1 polypeptide that is mutated at residue 186. The present invention also provides a STOP-1 variant polypeptide comprising STOP-1 that cannot disulfide bind with another STOP-1. According to one embodiment, the variant is a human STOP-1 polypeptide that is mutated at residue 55.
[0047] The present invention also provides nucleic acid molecules encoding the antibodies and polypeptides and variants thereof, vectors comprising the nucleic acid molecules, and host cells comprising the nucleic acid molecules of this invention.
[0048] The present invention includes compositions comprising an antibody, a polypeptide or a nucleic acid molecule of this invention. According to one embodiment, the composition further comprises a pharmaceutically acceptable carrier. In a further embodiment, the composition comprises a stromal targeting agent. In a further embodiment, the stromal targeting agent is covalently linked to the monoclonal antibody or polypeptide. In yet a further embodiment, the stromal targeting agent recognizes a stromal cell of a tumor.
[0049] The present invention provides methods for producing a STOP-1 polypeptide or an anti-STOP-1 antibody of this invention by culturing a cell comprising a nucleic acid according to this invention. According to one embodiment, the method for producing a STOP-1 polypeptide comprises the step of culturing a mammalian cell that comprises a nucleic acid molecule encoding the STOP-1 polypeptide and that is deficient in proteoglycan synthesis. According to another embodiment, the cell line that is deficient in proteoglycan synthesis is deficient in galactosyltransferase I activity. According to one preferred embodiment, the cell line is a CHO-psbg cell line.
[0050] The present invention provides a method for determining the presence of a STOP-1 polypeptide in a sample comprising exposing a sample suspected of containing the STOP-1 polypeptide to an anti-STOP-1 antibody and determining binding of said antibody to a component of said sample. According to one embodiment, the antibody is a monoclonal antibody of this invention.
[0051] The present invention provides methods for diagnosing or monitoring a cell proliferative disorder, such as a tumor, of a patient comprising the step of comparing the expression of STOP-1 in a normal tissue to the amount of STOP-1 being tested from the patient. In one embodiment, a STOP-1 protein can be detected by an agent such as an antibody of this invention. In another embodiment, STOP-1 mRNA can be detected by an agent such as a nucleic acid molecule that specifically hybridizes to the UNQ6762 mRNA. In a further embodiment, the tumor being tested has a large stromal compartment. In a further embodiment, STOP-1 detection agent is administered at or near the stromal compartment of the tissue being tested. In yet another embodiment, the method further comprises the step of observing or assaying the STOP-1 protein or mRNA in the stromal compartment agent of the normal tissue and tissue being tested. In another embodiment, the antibody is a monoclonal antibody of this invention.
[0052] The present invention provides a method of preventing or treating a proliferative disorder in a patient comprising the step of admininstering to the patient a composition of this invention in an amount effective to inhibit the proliferation of cells in the patient. In one embodiment, the proliferative disorder is desmoplasia. The present invention also provides a method of preventing or inhibiting the growth of a tumor that overexpresses STOP-1 in a patient comprising administering to the patient an antagonist of STOP-1 in an amount effective to inhibit growth of the tumor in the patient. In a further embodiment, the tumor to be treated has stromal compartments. In yet a further embodiment, the tumor having stromal compartments is selected from the group consisting of desmoid tumors, pancreatic cancer, sarcomas (e.g., hemangiosarcoma, rabdomyosarcoma) and adenocarcinomas (mammary adenocarcinomas, colon adenocarcinomas, gastrointestinal adenocarcinomas and ovarian adenocarcinomas), hepatocellular carcinoma, breast cancer, colon cancer, lung cancer, ovarian cancer, glioma, endometrial cancer and vascular cancer. In a further embodiment, the antagonist is administered at or near a stroma of the tumor. In another embodiment, the tumor is a melanoma or a round cell tumor (e.g., malignant fibrous hystiocytoma).
[0053] An antagonist according to this invention is any molecule that partially or fully blocks, inhibits, or neutralizes a biological activity of a native STOP-1 polypeptide and that specifically binds to a native STOP-1 polypeptide, wherein the binding of the antagonistic molecule (1) is to a native STOP-1 polypeptide in oligomeric form, (2) is to residues 94-243 of native human STOP-1 and/or (3) can be inhibited (e.g., as observed in a competitive ELISA assay using STOP-1 and 6B12) by a monoclonal antibody of this invention (e.g., a deposited antibody of this invention, etc.). According to one embodiment, the deposited antibody is the 6B12 antibody. According one embodiment, the antagonist is a polypeptide. According to another embodiment, the antagonist is an antibody of this invention. In another embodiment, the STOP-1 polypeptide that the antagonist inhibits is part of a trimeric complex.
[0054] According to another embodiment, the biological activity that is inhibited by the antagonist is the interaction of STOP-1 with a cell that specifically binds STOP-1. According to one embodiment, the cell is a breast cancer cell. According to another embodiment, the cell is an endothelial cell. According to yet another embodiment, the antagonist has an additional property selected from the group consisting of (1) capable of binding to an epitope within human STOP-1 that the 6B12 antibody binds; (2) capable of binding to a residue within at least residues 33-52 of human STOP-1; and (3) capable being competed from binding to STOP-1 by the 6B12 antibody (e.g., as observed in a competitive ELISA assay using STOP-1, the antagonist and the 6B12 antibody).
[0055] The present invention provides a method for inhibiting the growth of a cell that overexpresses STOP-1 comprising the step of inhibiting the secretion of STOP-1 from the cell. In one embodiment, secretion is inhibited by inhibiting glycosylation of STOP-1. In another embodiment, the secretion is inhibited by overexpressing a STOP-1 protein that cannot be secreted in the cell. In a further embodiment, the secretion is inhibited by a STOP-1 protein that is mutated at residue 186.
[0056] The present invention provides a method for preventing disulfide binding between STOP-1 molecules comprising a step selected from the group consisting of: (1) mutating STOP-1-encoding DNA molecules at residue cysteine 55; (2) expressing STOP-1 proteins that are mutated at residue cysteine 55 in the presence of naturally-occurring STOP-1 proteins; and (3) incubating STOP-1 proteins that are mutated at residue cysteine 55 with naturally-occurring STOP-1 proteins.
[0057] The present invention provides a method for cleaving STOP-1 comprising the step of incubating STOP-1 with a protease selected from the group consisting of a matrix metalloprotease-7 (MMP-7) and a matrix metalloprotease-9 (MMP-9). In a further embodiment, the method additionally comprises the step of monitoring the STOP-1 cleavage products produced.
[0058] Another embodiment of the present invention is directed to a method for inhibiting the growth of a cell that overexpresses a STOP-1 polypeptide, wherein the method comprises administering an antagonist of STOP-1, wherein the antagonist specifically binds to STOP-1 and is optionally conjugated to one or a combination of the agent(s) selected from the group consisting of a stromal targeting agent, a growth inhibitory agent or cytotoxic agent such as a toxin, including, for example, a maytansinoid or calicheamicin, an antibiotic, a radioactive isotope and a nucleolytic enzyme. Another embodiment of the present invention is directed to a method for inhibiting the growth of a cell that overexpresses a STOP-1 polypeptide, wherein the method comprises administering an agent to a stromal cell of the tumor, wherein the agent is an antagonist of STOP-1 or a nucleic acid molecule encoding a STOP polypeptide. The agent can be administered to the stromal cell directly by a patient or physician or indirectly, through the use of stromal targeting agents that can direct the agent to the stromal cell. The present invention provides an article of manufacture comprising (a) a composition of matter comprising a modified STOP-1 polypeptide, a STOP-1 polypeptide variant, STOP-1 antagonist, STOP-1 agonist, STOP-1 potentiator or a nucleic acid molecule encoding a STOP-1 polypeptide conjugated to a vehicle (e.g., such as antisense therapy or RNAi therapy); (b) a container containing said composition; and (c) a label affixed to said container, or a package insert included in said container referring to the use of said polypeptide variant, modified polypeptide or antagonist in the treatment of a proliferative disorder or a disease associated with abnormal angiogenesis or vasculogenesis (e.g., a package insert). According to one embodiment, the STOP-1 antagonist or potentiator is an antibody of this invention.
[0059] The present invention provides methods for testing the activity of STOP-1 and agonists or antagonists of STOP-1. In one embodiment, a method of inducing cell migration in vitro comprising administering to an endothelial cell a STOP-1 polypeptide in an amount effective to induce migration of said cell is provided. According to another embodiment, the present invention provides a method of testing the activity of a candidate antagonist or agonist of STOP-1 comprising the steps of treating a first endothelial cell with STOP-1, treating a second endothelial cell with STOP-1 and the candidate antagonist or agonist, and comparing the migration of the first and second endothelial cells. In one preferred embodiment, the cell used in such migration assay is a HUVEC cell.
[0060] The present invention also provides methods of treating a disease or condition associated with excessive, inappropriate or uncontrolled angiogenesis in a mammalian subject. In one embodiment, the method comprises the step of administering to the subject a STOP-1 antagonist in an amount effective to treat the disease, wherein the STOP-1 antagonist has any property selected from the group consisting of (1) binds to residues within human STOP-1 that the 6B12 antibody binds; (2) binds to a residue within at least residues 33-52 of human STOP-1; and (3) can be inhibited from binding to STOP-1 by the 6B12 antibody.
[0061] The present invention also contemplates treating patients who would benefit from increased angiogeneis vasculogenesis by administering a therapeutically effective amount of a STOP-1 potentiator, a molecule that enhances STOP-1 binding to cells and/or aggregates STOP-1 on the cell surface. Such a molecule would be administered in an amount effective to increase angiogenesis or vasculogenesis. In one preferred embodiment, the agonist is an anti-STOP-1 antibody that aggregates STOP-1 a cell surface.
[0062] The present invention also provides agonists comprising an oligomeric form of STOP-1 polypeptide that comprises greater than three STOP-1 polypeptides. According to one embodiment the agonist comprises six STOP-1 polypeptides. According to another embodiment, the STOP-1 polypeptide is part of an immunoadhesin that is used to form said agonist.
[0063] The present invention provides new methods for identifying and evaluating candidate and know STOP-1 antagonists, agonist and potentiators comprising the step of observing or measuring the binding of STOP-1 to a cell in the presence and absence of the antagonist, agonist or potentiator. According to one embodiment, the cell is a cancer cell. In a further embodiment the cell is a breast cancer cell. According to another embodiment, the cell is an endothelial cell.
BRIEF DESCRIPTION OF THE DRAWINGS
[0064] FIG. 1 shows an alignment of amino acid sequences encoding STOP-1 from a wide variety of species--human (SEQ ID NO:3), mouse (SEQ ID NO:4), rice fish (SEQ ID NO:5), zebrafish (SEQ ID NO:6) and chicken (SEQ ID NO:7). A consensus sequence is also provided. The arrow indicates a signal sequence cleavage site. Red indicates residues conserved in all species. Capitalized letters in the consensus sequence indicates residues that have been conserved throughout all species. Lower case letters in the consensus sequence indicate residues that are conserved in most species. Residues that that are not conserved in those species appear as a "period." "!" indicates I or V. "$" indicates L or M. "%" indicates F or Y. "#" indicates B, D, E, N, Q or Z.
[0065] FIG. 2 shows an amino acid sequence of human STOP-1 (SEQ ID NO:3). A signal sequence is indicated by the boxed amino acids. A triple helix domain is indicated by an underline. A glycosylation site is at amino acid 186.
[0066] FIG. 3 shows (A) the presence of human STOP-1 mRNA in certain tissue types and (B) mouse STOP-1 mRNA from different stages of mouse development. Full length human or mouse STOP-1 DNAs were radiolabelled and used to probe northern blots of tissues from adult humans or developing mouse embryos.
[0067] FIG. 4 shows coomassie-stained human STOP-1 proteins produced by (A) CHO-DP12 or (B) CHO-psgb (ATCC) cells and purified by nickel-NTA affinity chromatography. The vector, pRK5, was used as a control.
[0068] FIG. 5 shows a western blot of human histidine-tagged STOP-1 protein present in the (A) supernatant and (B) cell lysate of transient transfected CHO-psgb cells. The western blot was probed with anti-his antibody.
[0069] FIG. 6 shows the oligomerization of human STOP-1 protein expressed using a baculoviral infection system in SF9 insect cells. STOP-1 protein and various deletion mutants were expressed from SF9 cells, separated on a size exclusion column and subjected to light scattering analysis e.g., (A) S31-K243, (B) E89-K243 and (C) L94-K243. The predicted molecular weight of the monomers appear in the left corner of each graph. The numbers appearing next to several peaks refer to the average molecular weight of the complexes in the peak
[0070] FIG. 7 shows the oligomerization of human STOP-1 protein expressed from mammalian cells. Human STOP-1 protein and various deletion mutants were expressed from CHO cells, separated on a size exclusion column and subjected to light scattering analysis, e.g., (A) M1-K243 and (B) delta-THD (residues 1-54, 94-243, plus histidine tag). The predicted molecular weight of the monomers appear in the left corner of each graph. The numbers appearing next to several peaks refer to an average molecular weight of the complexes in those peaks. Under non-reducing conditions, western blots of secreted full length, his-tagged human STOP-1 protein recombinantly expressed from CHO-psgb cells presented predominantly homodimerized complexes (C). The western blots were probed with anti-his antibody.
[0071] FIG. 8 shows western blots of (A) cell culture media and (B) whole cell lysates from CHO-psgb cells expressing human his-tagged STOP-1 WT, delta-THD, and delta-delta-THD STOP-1 (residues 1-51, 94-243, plus histidine tag) and subjected to reducing or non-reducing conditions. The western blots were probed with anti-his antibody.
[0072] FIG. 9 shows western blots of (A) cell culture media and (B) whole cell lysates from CHO-psgb cells expressing his-tagged WT, G53A and N186A STOP-1 constructs and subjected to reducing or non-reducing conditions. The western blots were probed with anti-his antibody.
[0073] FIG. 10 shows western blots of (A) cell culture media and (B) whole cell lysates from CHO-psgb cells expressing his-tagged WT, C55A, C93A and C109A STOP-1 constructs and subjected to reducing or non-reducing conditions. The western blots were probed with anti-his antibody.
[0074] FIG. 11 shows that murine STOP-1 mRNA (mSTOP-1 mRNA) is expressed in breast tumors derived from MMTV-WNT1 transgenic mice but not in normal mammary epithelial cells. RNA samples were taken from breast tumor cells (marked "T1"-"T7") or C57 Mg mouse normal mammary epithelial cells (marked, "N"), subjected to RT-PCR with mSTOP-1 primers and mRLP19 primers. The PCR products were separated on an agarose gel.
[0075] FIG. 12 shows the proliferation of 3T3 cells after transfection with (A) human STOP-1 or (B) mouse STOP-1. FIG. 12A shows the amount of 3H-thymidine incorporation (counts per minute (cpm)) in 3T3 cells at 12, 28 and 96 hours after addition of 41-thymidine. FIG. 12B shows the amount of 3H-thymidine incorporation (counts per minute (cpm)) in 3T3 cells at 12, 28 and 96 hours after addition of 3H-thymidine. Controls: transfections with vector alone (puro2 and ph1).
[0076] FIG. 13 shows the proliferation of 3T3 or 293 cells after infection with retrovirus encoding human STOP-1. FIGS. 13A and B are western blots of human STOP-1 proteins expressed from 3T3 cells or 293, respectively, infected with a retrovirus encoding a control vector (Babe) or human STOP-1. STOP-1 was immunoprecipitated from whole cell lysates using the S7-IgG antibody. Western blots were probed with polyclonal anti-human STOP-1 antibodies. FIG. 13C shows the level of cell proliferation observed for the infected cell populations as detected by a colorimetric Cell Titer Assay.
[0077] FIG. 14 shows that mouse STOP-1 promotes tumorigenesis by 3T3 fibroblasts in a xenograft mouse model. FIG. 14A shows the mean volume of tumors in mice implanted with 3T3 fibroblasts transfected with vector alone (p2 vector) or DNA encoding mouse STOP-1 or RAS protein. The transfected cells were implanted into nude mice or tested for protein expression. FIGS. 14B and C show western blots of aliquots of the supernatants and lysates, respectively, of the transfected cells. The western blot was probed with rabbit anti-STOP-1 polyclonal antibodies. "TI" refers to the tumor incident ratio.
[0078] FIG. 15 shows that human STOP-1 promotes tumorigenesis by 3T3 fibroblasts in a xenograft mouse model. FIG. 15 shows the mean tumor volume of tumors in mice implanted with 3T3 fibroblasts transfected with vector alone or DNA encoding human STOP-1, RAS protein or LP1.
[0079] FIG. 16 shows that recombinant STOP-1 protein potentiates SK-Mel-31 cells wound healing and motility. SK-Mel-31 cells were treated with (A) NT--no exogenous ligand treatment, (B) b762--baculoviral produced human STOP-1 protein, (C) hrEGF--(50 ng/ml), (D) hrEGF and b762 or (E) CHO mammalian produced human STOP-1 protein.
[0080] FIG. 17 shows that anti-human STOP-1 antibody, 6B12, binds to the N-terminal sequence of human STOP-1 between the signal sequence and triple helix domains FIG. 17A is a schematic of the his-tagged human and full length zebrafish STOP-1 proteins used in the epitope location studies of FIGS. 18B and C. FIGS. 17B and C show western blots probed with anti-his antibody and 6B12 antibody, respectively, of extracts from cells that recombinantly expressed the proteins of FIG. 17A.
[0081] FIG. 18 shows the amino acid sequences of the CDRs of several phage-derived antibodies having affinity for human STOP-1. "H1," "H2" and "H3" refer to VH-CDR1, VH-CDR2 and VH-CDR3. The numerical header generally corresponds to amino acid positions 28-33, 49-58 and 92-102 according to the Kabat numbering system. The SEQ ID NOs for the listed sequences are as follows:
TABLE-US-00004 H1 H2 H3 Ab Name SEQ ID NO: 8 SEQ ID NO: 9 SEQ ID NO: 10 S7 SEQ ID NO: 11 SEQ ID NO: 12 SEQ ID NO: 13 S16 SEQ ID NO: 14 SEQ ID NO: 15 SEQ ID NO: 16 F5, F6 SEQ ID NO: 17 SEQ ID NO: 18 SEQ ID NO: 19 S4, F13, F37 SEQ ID NO: 20 SEQ ID NO: 21 SEQ ID NO: 22 S9
[0082] FIG. 19 shows a graph of an ELISA assay to determine an optimal concentration of S4 and S7 Fab or IgG for use in a competition ELISA to determine the affinity of the antibodies for STOP-1. "S coated" refers to a short form (#94-243) of STOP-1 coated on a microtiter plate. "F coated" refers to a full-length form of human STOP-1 coated on a microtiter plate. Approximately 90% of maximal binding was considered to be optimal for use in a competitive ELISA assay. Horse-radish peroxidase-conjugated protein G was used to detect the bound Fab and IgG.
[0083] FIG. 20 shows a graph of the results of competitive ELISA to determine the binding affinities of the S4 and S7 Fab or IgG. The plates were coated with short form or full-length human STOP-1 and competed with short form or full length STOP-1, respectively (FIGS. 20A and B, respectively). The calculated binding affinities are indicated in the parentheticals.
[0084] FIG. 21 shows a summary of the binding affinities of several phage-derived antibodies against STOP-1. "S/S" refers to an ELISA in which the microtiter plate was coated with a short form of STOP-1 and competed with a short form of STOP-1. "F/S" refers to an ELISA in which the microtiter plate was coated with a full-length form of human STOP-1 and competed with a short form of human STOP-1. "F/F" refers to an ELISA in which the microtiter plate was coated with a full-length form of STOP-1 and competed with a full-length form of STOP-1. The phage used in these studies were the S4-Fab phage and the S7-F(ab)'2 phage.
[0085] FIG. 22 shows a graph of an ELISA assay in which the plates were coated with human STOP-1, bound with S4 IgG and then competed with S4 (Fab) phage, S7 (F(ab)'2) phage, S9 (Fab) phage, S16 (F(ab)'2) phage and F5 (F(ab)'2) phage. The Y axis refers to percentage unblocked as calculated by dividing the OD450 nm value of the well that blocked S4 IgG by the OD450 nm value of a well without S4 IgG.
[0086] FIG. 23 shows a coomassie stained gel of baculovirus-expressed human STOP-1 protein cleaved by various proteases in vitro. "MMP" refers to matrix metalloprotease.
[0087] FIG. 24A-D are schematics of phagemids encoding Fab of F(ab)'2 phage display proteins or vectors encoding Fab or IgG proteins. FIG. 24A is a schematic of a Fab-phagemid construct. The construct contains an alkaline phosphatase promoter, an STII signal sequence, a VL and CL light chain sequence, a gD tag, another STII signal sequence, a heavy chain VH and CH1 region and a C-terminal part of the M13 bacteriophage pIII coat protein (cP3). FIG. 24B is a schematic of a F(ab)'2-phagemid construct. The construct contains generally the same sequences as the Fab-phagemid, except it additionally includes a leucine zipper sequence (Zip). FIG. 24C is a schematic of a nucleic acid molecule encoding a Fab protein. FIG. 24D is a schematic of a nucleic acid molecule encoding an IgG protein, which IgG protein includes a CH2 and CH3 sequence.
[0088] FIG. 25A-H describe amino acid sequences and a nucleic acid sequence for a phage display anti-Her-2 Fab. More specifically, FIG. 25 shows an amino acid sequence comprising an anti-Her-2 Fab light chain (SEQ ID NO:86), an amino acid sequence comprising an anti-Her-2 Fab heavy chain region (SEQ ID NO:87) and the nucleic acid sequence of a phagemid encoding the amino acid sequences (SEQ ID NO:88).
[0089] FIG. 26A-H describe amino acid sequences and a nucleic acid sequence for a phage display anti-Her-2 F(ab)'2. More specifically, FIG. 26 shows an amino acid sequence comprising an anti-Her-2 F(ab)'2 light chain (SEQ ID NO:89), an amino acid sequence comprising an anti-Her-2 F(ab)'2 heavy chain region (SEQ ID NO:90) and the nucleic acid sequence of a phagemid encoding the amino acid sequences (SEQ ID NO:91).
[0090] FIG. 27A-C describe amino acid sequences and a nucleic acid sequence for a phage display S4-Fab. More specifically, FIG. 27 shows an amino acid sequence comprising an S4-Fab light chain (SEQ ID NO:92), an amino acid sequence comprising an S4-Fab heavy chain region (SEQ ID NO:93) and a nucleic acid sequence encoding the amino acid sequences (SEQ ID NO:94).
[0091] FIG. 28A-C describe amino acid sequences and a nucleic acid sequence for a phage display S9 Fab. More specifically, FIG. 28 shows an amino acid sequence comprising an S9-Fab light chain (SEQ ID NO:95), an amino acid sequence comprising an S9-Fab heavy chain region (SEQ ID NO:96) and a nucleic acid sequence encoding the amino acid sequences (SEQ ID NO:97).
[0092] FIG. 29A-C describe amino acid sequences and a nucleic acid sequence for a phage display S7-F(ab)'2. More specifically, FIG. 29 shows an amino acid sequence comprising an S7-F(ab)'2 light chain (SEQ ID NO:98), an amino acid sequence comprising an S7-F(ab)'2 heavy chain region (SEQ ID NO:99) and a nucleic acid sequence encoding the amino acid sequences (SEQ ID NO:100).
[0093] FIG. 30A-C describe amino acid sequences and a nucleic acid sequence for a phage display 516-F(ab)'2. More specifically, FIG. 30 shows an amino acid sequence comprising an 516-F(ab)'2 light chain (SEQ ID NO:101), an amino acid sequence comprising an S16-F(ab)'2 heavy chain region (SEQ ID NO:102) and a nucleic acid sequence encoding the amino acid sequences (SEQ ID NO:103).
[0094] FIG. 31A-C describe amino acid sequences and a nucleic acid sequence for a phage display F5-F(ab)'2. FIG. 31 shows an amino acid sequence comprising a F5-F(ab)'2 light chain (SEQ ID NO:104), an amino acid sequence comprising an F5-F(ab)'2 heavy chain region (SEQ ID NO:105) and a nucleic acid sequence encoding the amino acid sequences (SEQ ID NO:106).
[0095] FIG. 32A-G describe amino acid sequences and a nucleic acid sequence for a S4-Fab. More specifically, FIG. 32 shows an amino acid sequence comprising an S4-Fab light chain (SEQ ID NO:107), an amino acid sequence comprising an S4-Fab heavy chain region (SEQ ID NO:108) and the nucleic acid sequence of a vector encoding the amino acid sequence (SEQ ID NO:109).
[0096] FIG. 33A-F describe an S4 light chain sequence of an IgG protein. More specifically, FIG. 33 shows an amino acid sequence comprising an S4 Light Chain (SEQ ID NO:110) and the nucleic acid sequence of a vector encoding the amino acid sequence (SEQ ID NO:111).
[0097] FIG. 34A-G describe an S4 heavy chain sequence of an IgG protein. More specifically, FIG. 34 shows an amino acid sequence comprising an S4 Heavy Chain (SEQ ID NO:112) and the nucleic acid sequence of a vector encoding the amino acid sequence (SEQ ID NO:113).
[0098] FIG. 35 shows a frequency of amino acids in human antibody light chain sequences from the Kabat database.
[0099] FIG. 36 shows one illustrative embodiment of a suitable codon set design.
[0100] FIG. 37 is an illustrative embodiment of restricted diversity degenerate (also referred to herein as "nonrandom") codon sets for diversification of CDRs L1, L2 & L3.
[0101] FIG. 38 is an illustrative embodiment of restricted diversity degenerate (also referred to herein as "nonrandom") codon sets for diversification of CDRs L1, L2 & L3.
[0102] FIG. 39 is an illustrative embodiment of restricted diversity degenerate (also referred to herein as "nonrandom") codon sets for diversification of CDR L3.
[0103] FIG. 40 is an illustrative embodiment of a restricted diversity degenerate (also referred to herein as "nonrandom") codon sets for diversification of CDRs L1, L2 & L3.
[0104] FIG. 41 shows a flow cytometric analysis of populations of 293, HeLa, HT1080 or HUVEC cells treated with either (1) anti-HIS antibodies, (2) anti-HIS antibodies and STOP-1 protein or (3) anti-flag antibodies and STOP-1 protein, followed by treatment with fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse antibodies. A small, insignificant number of cells bound the anti-flag antibodies (i.e., the peaks at far left corner of the x-axis in the 293, HeLa and HT1080 graphs). The x-axis indicates the number of cells (log fluorescein signal intensity). The y-axis indicates the level of fluorescence emitted by the labeled cells (events).
[0105] FIG. 42 shows a FACS analysis of populations of HT1080 cells treated with (1) anti-HIS antibodies, (2) anti-HIS antibodies and STOP-1 protein, (3) anti-flag antibodies and STOP-1 protein, (3) STOP-1 protein, (4) STOP-1 protein and S7 antibodies or (5) STOP-1 and 6b12 antibodies, followed by treatment with FITC-conjugated goat anti-mouse antibodies.
[0106] FIG. 43 charts the migration of HT1080 cells (number of cells) in a modified Boyden chemotactic chamber after treatment with bFGF or STOP-1 ("762") or a negative control.
[0107] FIG. 44 shows a FACS analysis of STOP-1 binding to MDA435 cells in the presence and absence of an anti-STOP-1 antibody (6B12) or an antibody control (4B7). The detection antibody, anti-flag M2-FITC antibody, did not effect STOP-1 binding.
[0108] FIG. 45 is a graph that shows the fold change in STOP-1 mRNA expression after treatment (A) under hypoxic conditions for 8 and 34 hours or (B) under normoxic conditions for 3, 8 and 34 hours, in the presence and absence of recombinant human TNFalpha.
DETAILED DESCRIPTION OF THE INVENTION
[0109] A nucleic acid sequence coding for a STOP-1 protein according to this invention includes, e.g., SEQ ID NO:1 and the nucleic acid molecules encoding the polypeptides of FIG. 1.
TABLE-US-00005 SEQ ID NO: 1 GGAGAGAGGCGCGCGGGTGAAAGGCGCATTGATGCAGCCTGCGGCGGC CTCGGAGCGCGGCGGAGCCAGACGCTGACCACGTTCCTCTCCTCGGTC TCCTCCGCCTCCAGCTCCGCGCTGCCCGGCAGCCGGGAGCCATGCGAC CCCAGGGCCCCGCCGCCTCCCCGCAGCGGCTCCGCGGCCTCCTGCTGC TCCTGCTGCTGCAGCTGCCCGCGCCGTCGAGCGCCTCTGAGATCCCCA AGGGGAAGCAAAAGGCGCAGCTCCGGCAGAGGGAGGTGGTGGACCTGT ATAATGGAATGTGCTTACAAGGGCCAGCAGGAGTGCCTGGTCGAGACG GGAGCCCTGGGGCCAATGTTATTCCGGGTACACCTGGGATCCCAGGTC GGGATGGATTCAAAGGAGAAAAGGGGGAATGTCTGAGGGAAAGCTTTG AGGAGTCCTGGACACCCAACTACAAGCAGTGTTCATGGAGTTCATTGA ATTATGGCATAGATCTTGGGAAAATTGCGGAGTGTACATTTACAAAGA TGCGTTCAAATAGTGCTCTAAGAGTTTTGTTCAGTGGCTCACTTCGGC TAAAATGCAGAAATGCATGCTGTCAGCGTTGGTATTTCACATTCAATG GAGCTGAATGTTCAGGACCTCTTCCCATTGAAGCTATAATTTATTTGG ACCAAGGAAGCCCTGAAATGAATTCAACAATTAATATTCATCGCACTT CTTCTGTGGAAGGACTTTGTGAAGGAATTGGTGCTGGATTAGTGGATG TTGCTATCTGGGTTGGCACTTGTTCAGATTACCCAAAAGGAGATGCTT CTACTGGATGGAATTCAGTTTCTCGCATCATTATTGAAGAACTACCAA AATAAATGCTTTAATTTTCATTTGCTACCTCTTTTTTTATTATGCCTT GGAATGGTTCACTTAAATGACATTTTAAATAAGTTTATGTATACATCT GAATGAAAAGCAAAGCTAAATATGTTTACAGACCAAAGTGTGATTTCA CACTGTTTTTAAATCTAGCATTATTCATTTTGCTTCAATCAAAAGTGG TTTCAATATTTTTTTTAGTTGGTTAGAATACTTTCTTCATAGTCACAT TCTCTCAACCTATAATTTGGAATATTGTTGTGGTCTTTTGTTTTTTCT CTTAGTATAGCATTTTTAAAAAAATATAAAAGCTACCAATCTTTGTAC AATTTGTAAATGTTAAGAATTTTTTTTATATCTGTTAAATAAAAATTA TTTCCAACA
[0110] The terms "STOP-1," "STOP-1 protein," "STOP-1 polypeptide" (also referred to UNQ762 or 762) as used herein include native sequence polypeptides, polypeptide variants and fragments of native sequence polypeptides and polypeptide variants (which are further defined herein), unless specified otherwise. STOP-1 proteins can be obtained from various species, e.g., humans, by using antibodies according to this invention or by recombinant or synthetic methods, including using deposited nucleic acid molecules. An oligomeric form of STOP-1 includes a human STOP-1 having only residues 94-243, or a part thereof. An oligomeric form according to this invention can include a dimer, a trimer and a hexamer of STOP-1. According to one preferred embodiment, the oligomeric form of STOP-1 is a trimer.
[0111] A "native sequence" polypeptide or "native" polypeptide is one which has the same amino acid sequence as a polypeptide (e.g., antibody) derived from nature. A "native sequence" polypeptide is one which has the same amino acid sequence as a polypeptide (e.g., antibody) derived from nature. Such native sequence polypeptides can be isolated from nature or can be produced by recombinant or synthetic means. Thus, a native sequence polypeptide can have the amino acid sequence of a naturally occurring human polypeptide, murine polypeptide, or polypeptide from any other mammalian species. A "native sequence" STOP-1 polypeptide or a "native" STOP-1 polypeptide comprises a polypeptide having the same amino acid sequence as the corresponding STOP-1 polypeptide derived from nature. For example, in one preferred embodiment, the nucleic acid sequence encoding a native sequence of human STOP-1 can be found in SEQ ID NO:2 and FIG. 2.
TABLE-US-00006 SEQ ID NO: 2 MRPQGPAASPQRLRGLLLLLLLQLPAPSSASEIPKGKQKAQLRQREVV DLYNGMCLQGPAGVPGRDGSPGANVIPGTPGIPGRDGFKGEKGECLRE SFEESWTPNYKQCSWSSLNYGIDLGKIAECTFTKMRSNSALRVLFSGS LRLKCRNACCQRWYFTFNGAECSGPLPIEAIIYLDQGSPEMNSTINIH RTSSVEGLCEGIGAGLVDVAIWVGTCSDYPKGDASTGWNSVSRIIIEE LPK
[0112] Such STOP-1 polypeptides can be isolated from nature or can be produced by recombinant or synthetic means. The term "native sequence" or "native" STOP-1 polypeptide or protein specifically encompasses naturally-occurring truncated or secreted forms of the STOP-1 protein, naturally-occurring variant forms (e.g., alternatively spliced forms) and naturally-occurring allelic variants of the polypeptide. In certain embodiments of the invention, the native sequence STOP-1 polypeptides disclosed herein are mature or full-length native sequence polypeptides comprising the full-length amino acids sequences shown in the accompanying figures.
[0113] The approximate location of the "signal peptides" of the various STOP-1 polypeptides disclosed herein can be seen in the present specification and/or the accompanying figures. It is also recognized that, in some cases, cleavage of a signal sequence from a secreted polypeptide is not entirely uniform, resulting in more than one secreted species. These mature polypeptides, where the signal peptide is cleaved within no more than about 5 amino acids on either side of the C-terminal boundary of the signal peptide as identified herein, and the polynucleotides encoding them, are contemplated by the present invention.
[0114] "STOP-1 polypeptide variant" means a STOP-1 polypeptide having at least about 80% amino acid sequence identity with a full-length native sequence STOP-1 polypeptide sequence as disclosed herein, a STOP-1 polypeptide sequence lacking the signal peptide or triple helix domain as disclosed herein, or any other fragment of a full-length STOP-1 polypeptide sequence as disclosed herein (such as those encoded by a nucleic acid that represents only a portion of the complete coding sequence for a full-length STOP-1 polypeptide). Such STOP-1 polypeptide variants include, for instance, STOP-1 polypeptides wherein one or more amino acid residues are added, or deleted, at the N- or C-terminus of the full-length native amino acid sequence. Ordinarily, a STOP-1 polypeptide variant will have at least about 80% amino acid sequence identity, alternatively at least about 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% amino acid sequence identity, to a full-length native sequence STOP-1 polypeptide sequence as disclosed herein, a STOP-1 polypeptide sequence lacking the signal peptide as disclosed herein, a triple helix domain of a STOP-1 polypeptide, with or without the signal peptide, as disclosed herein or any other specifically defined fragment of a full-length STOP-1 polypeptide sequence as disclosed herein. Ordinarily, STOP-1 variant polypeptides are at least about 10 amino acids in length, alternatively at least about 20, 30, 40, 50, 60, 70, 80, 90, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290, 300, 310, 320, 330, 340, 350, 360, 370, 380, 390, 400, 410, 420, 430, 440, 450, 460, 470, 480, 490, 500, 510, 520, 530, 540, 550, 560, 570, 580, 590, 600 amino acids in length, or more. Optionally, STOP-1 variant polypeptides will have no more than one conservative amino acid substitution as compared to the native STOP-1 polypeptide sequence, alternatively no more than 2, 3, 4, 5, 6, 7, 8, 9, or 10 conservative amino acid substitution as compared to the native STOP-1 polypeptide sequence.
[0115] "Percent (%) amino acid sequence identity" with respect to the STOP-1 polypeptide sequences identified herein is defined as the percentage of amino acid residues in a candidate sequence that are identical with the amino acid residues in the specific STOP-1 polypeptide sequence, after aligning the sequences and introducing gaps, if necessary, to achieve the maximum percent sequence identity, and not considering any conservative substitutions as part of the sequence identity. Alignment for purposes of determining percent amino acid sequence identity can be achieved in various ways that are within the skill in the art, for instance, using publicly available computer software such as BLAST, BLAST-2, ALIGN or Megalign (DNASTAR) software. Those skilled in the art can determine appropriate parameters for measuring alignment, including any algorithms needed to achieve maximal alignment over the full length of the sequences being compared. For purposes herein, however, % amino acid sequence identity values are generated using the sequence comparison computer program ALIGN-2, wherein the complete source code for the ALIGN-2 program is provided in Table 1 below. The ALIGN-2 sequence comparison computer program was authored by Genentech, Inc. and the source code shown in Table 1 below has been filed with user documentation in the U.S. Copyright Office, Washington D.C., 20559, where it is registered under U.S. Copyright Registration No. TXU510087. The ALIGN-2 program is publicly available through Genentech, Inc., South San Francisco, Calif. or can be compiled from the source code provided in Table 1 below. The ALIGN-2 program should be compiled for use on a UNIX operating system, preferably digital UNIX V4.0D. All sequence comparison parameters are set by the ALIGN-2 program and do not vary.
[0116] In situations where ALIGN-2 is employed for amino acid sequence comparisons, the % amino acid sequence identity of a given amino acid sequence A to, with, or against a given amino acid sequence B (which can alternatively be phrased as a given amino acid sequence A that has or comprises a certain % amino acid sequence identity to, with, or against a given amino acid sequence B) is calculated as follows:
100 times the fraction X/Y
[0117] where X is the number of amino acid residues scored as identical matches by the sequence alignment program ALIGN-2 in that program's alignment of A and B, and where Y is the total number of amino acid residues in B. It will be appreciated that where the length of amino acid sequence A is not equal to the length of amino acid sequence B, the % amino acid sequence identity of A to B will not equal the % amino acid sequence identity of B to A. As examples of % amino acid sequence identity calculations using this method, Tables 2 and 3 demonstrate how to calculate the % amino acid sequence identity of the amino acid sequence designated "Comparison Protein" to the amino acid sequence designated "STOP-1", wherein "STOP-1" represents the amino acid sequence of a hypothetical STOP-1 polypeptide of interest, "Comparison Protein" represents the amino acid sequence of a polypeptide against which the "STOP-1" polypeptide of interest is being compared, and "X, "Y" and "Z" each represent different hypothetical amino acid residues. Unless specifically stated otherwise, all % amino acid sequence identity values used herein are obtained as described in the immediately preceding paragraph using the ALIGN-2 computer program.
[0118] "STOP-1 variant polynucleotide" or "STOP-1 variant nucleic acid sequence" means a nucleic acid molecule which encodes a STOP-1 polypeptide, preferably an active STOP-1 polypeptide, as defined herein and which has at least about 80% nucleic acid sequence identity with a nucleotide acid sequence encoding a full-length native sequence STOP-1 polypeptide sequence as disclosed herein, a full-length native sequence STOP-1 polypeptide sequence lacking the signal peptide as disclosed herein, the triple helix domain of a STOP-1 polypeptide, with or without the signal peptide, as disclosed herein or any other fragment of a full-length STOP-1 polypeptide sequence as disclosed herein (e.g., residues 94-243 of human STOP-1). Ordinarily, a STOP-1 variant polynucleotide will have at least about 80% nucleic acid sequence identity, alternatively at least about 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% nucleic acid sequence identity with a nucleic acid sequence encoding a full-length native sequence STOP-1 polypeptide sequence as disclosed herein, a full-length native sequence STOP-1 polypeptide sequence lacking the signal peptide as disclosed herein, the triple helix domain of a STOP-1 polypeptide, with or without the signal sequence, as disclosed herein or any other fragment of a full-length STOP-1 polypeptide sequence as disclosed herein. Variants do not encompass the native nucleotide sequence.
[0119] Ordinarily, STOP-1 variant polynucleotides are at least about 5 nucleotides in length, alternatively at least about 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 105, 110, 115, 120, 125, 130, 135, 140, 145, 150, 155, 160, 165, 170, 175, 180, 185, 190, 195, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290, 300, 310, 320, 330, 340, 350, 360, 370, 380, 390, 400, 410, 420, 430, 440, 450, 460, 470, 480, 490, 500, 510, 520, 530, 540, 550, 560, 570, 580, 590, 600, 610, 620, 630, 640, 650, 660, 670, 680, 690, 700, 710, 720, 730, 740, 750, 760, 770, 780, 790, 800, 810, 820, 830, 840, 850, 860, 870, 880, 890, 900, 910, 920, 930, 940, 950, 960, 970, 980, 990, or 1000 nucleotides in length, wherein in this context the term "about" means the referenced nucleotide sequence length plus or minus 10% of that referenced length.
[0120] "Percent (%) nucleic acid sequence identity" with respect to STOP-1-encoding nucleic acid sequences identified herein is defined as the percentage of nucleotides in a candidate sequence that are identical with the nucleotides in the STOP-1 nucleic acid sequence of interest, after aligning the sequences and introducing gaps, if necessary, to achieve the maximum percent sequence identity. Alignment for purposes of determining percent nucleic acid sequence identity can be achieved in various ways that are within the skill in the art, for instance, using publicly available computer software such as BLAST, BLAST-2, ALIGN or Megalign (DNASTAR) software. For purposes herein, however, % nucleic acid sequence identity values are generated using the sequence comparison computer program ALIGN-2, wherein the complete source code for the ALIGN-2 program is provided in Table 1 below. The ALIGN-2 sequence comparison computer program was authored by Genentech, Inc. and the source code shown in Table 1 below has been filed with user documentation in the U.S. Copyright Office, Washington D.C., 20559, where it is registered under U.S. Copyright Registration No. TXU510087. The ALIGN-2 program is publicly available through Genentech, Inc., South San Francisco, Calif. or can be compiled from the source code provided in Table 1 below. The ALIGN-2 program should be compiled for use on a UNIX operating system, preferably digital UNIX V4.0D. All sequence comparison parameters are set by the ALIGN-2 program and do not vary.
[0121] In situations where ALIGN-2 is employed for nucleic acid sequence comparisons, the % nucleic acid sequence identity of a given nucleic acid sequence C to, with, or against a given nucleic acid sequence D (which can alternatively be phrased as a given nucleic acid sequence C that has or comprises a certain % nucleic acid sequence identity to, with, or against a given nucleic acid sequence D) is calculated as follows:
100 times the fraction W/Z
[0122] where W is the number of nucleotides scored as identical matches by the sequence alignment program ALIGN-2 in that program's alignment of C and D, and where Z is the total number of nucleotides in D. It will be appreciated that where the length of nucleic acid sequence C is not equal to the length of nucleic acid sequence D, the % nucleic acid sequence identity of C to D will not equal the % nucleic acid sequence identity of D to C. As examples of % nucleic acid sequence identity calculations, Tables 4 and 5, demonstrate how to calculate the % nucleic acid sequence identity of the nucleic acid sequence designated "Comparison DNA" to the nucleic acid sequence designated "STOP-1-DNA", wherein "STOP-1-DNA" represents a hypothetical STOP-1-encoding nucleic acid sequence of interest, "Comparison DNA" represents the nucleotide sequence of a nucleic acid molecule against which the "STOP-1-DNA" nucleic acid molecule of interest is being compared, and "N", "L" and "V" each represent different hypothetical nucleotides. Unless specifically stated otherwise, all % nucleic acid sequence identity values used herein are obtained as described in the immediately preceding paragraph using the ALIGN-2 computer program.
[0123] In other embodiments, STOP-1 variant polynucleotides are nucleic acid molecules that encode a STOP-1 polypeptide and which are capable of hybridizing, preferably under stringent hybridization and wash conditions, to nucleotide sequences encoding a full-length STOP-1 polypeptide as disclosed herein. STOP-1 variant polypeptides can be those that are encoded by a STOP-1 variant polynucleotide.
[0124] The term "full-length coding region" when used in reference to a nucleic acid encoding a STOP-1 polypeptide refers to the sequence of nucleotides which encode the full-length STOP-1 polypeptide of the invention (which is often shown between start and stop codons, inclusive thereof, in the accompanying figures). The term "full-length coding region" when used in reference to an ATCC deposited nucleic acid refers to the STOP-1 polypeptide-encoding portion of the cDNA that is inserted into the vector deposited with the ATCC (which is often shown between start and stop codons, inclusive thereof, in the accompanying figures).
[0125] "Isolated," when used to describe the various STOP-1 polypeptides disclosed herein, means polypeptide that has been identified and separated and/or recovered from a component of its natural environment. Contaminant components of its natural environment are materials that would typically interfere with diagnostic or therapeutic uses for the polypeptide, and can include enzymes, hormones, and other proteinaceous or non-proteinaceous solutes. In preferred embodiments, the polypeptide will be purified (1) to a degree sufficient to obtain at least 15 residues of N-terminal or internal amino acid sequence by use of a spinning cup sequenator, or (2) to homogeneity by SDS-PAGE under non-reducing or reducing conditions using Coomassie blue or, preferably, silver stain. Isolated polypeptide includes polypeptide in situ within recombinant cells, since at least one component of the STOP-1 polypeptide natural environment will not be present. Ordinarily, however, isolated polypeptide will be prepared by at least one purification step.
[0126] An "isolated" STOP-1 polypeptide-encoding nucleic acid or other polypeptide-encoding nucleic acid is a nucleic acid molecule that is identified and separated from at least one contaminant nucleic acid molecule with which it is ordinarily associated in the natural source of the polypeptide-encoding nucleic acid. An isolated polypeptide-encoding nucleic acid molecule is other than in the form or setting in which it is found in nature. Isolated polypeptide-encoding nucleic acid molecules therefore are distinguished from the specific polypeptide-encoding nucleic acid molecule as it exists in natural cells. However, an isolated polypeptide-encoding nucleic acid molecule includes polypeptide-encoding nucleic acid molecules contained in cells that ordinarily express the polypeptide where, for example, the nucleic acid molecule is in a chromosomal location different from that of natural cells.
[0127] The term "control sequences" refers to DNA sequences necessary for the expression of an operably linked coding sequence in a particular host organism. The control sequences that are suitable for prokaryotes, for example, include a promoter, optionally an operator sequence, and a ribosome binding site. Eukaryotic cells are known to utilize promoters, polyadenylation signals, and enhancers.
[0128] Nucleic acid is "operably linked" when it is placed into a functional relationship with another nucleic acid sequence. For example, DNA for a presequence or secretory leader is operably linked to DNA for a polypeptide if it is expressed as a preprotein that participates in the secretion of the polypeptide; a promoter or enhancer is operably linked to a coding sequence if it affects the transcription of the sequence; or a ribosome binding site is operably linked to a coding sequence if it is positioned so as to facilitate translation. Generally, "operably linked" means that the DNA sequences being linked are contiguous, and, in the case of a secretory leader, contiguous and in reading phase. However, enhancers do not have to be contiguous. Linking is accomplished by ligation at convenient restriction sites. If such sites do not exist, the synthetic oligonucleotide adaptors or linkers are used in accordance with conventional practice.
[0129] "Stringency" of hybridization reactions is readily determinable by one of ordinary skill in the art, and generally is an empirical calculation dependent upon probe length, washing temperature, and salt concentration. In general, longer probes require higher temperatures for proper annealing, while shorter probes need lower temperatures. Hybridization generally depends on the ability of denatured DNA to reanneal when complementary strands are present in an environment below their melting temperature. The higher the degree of desired homology between the probe and hybridizable sequence, the higher the relative temperature which can be used. As a result, it follows that higher relative temperatures would tend to make the reaction conditions more stringent, while lower temperatures less so. For additional details and explanation of stringency of hybridization reactions, see Ausubel et al., Current Protocols in Molecular Biology, Wiley Interscience Publishers, (1995).
[0130] "Stringent conditions" or "high stringency conditions", as defined herein, can be identified by those that: (1) employ low ionic strength and high temperature for washing, for example 0.015 M sodium chloride/0.0015 M sodium citrate/0.1% sodium dodecyl sulfate at 50 C; (2) employ during hybridization a denaturing agent, such as formamide, for example, 50% (v/v) formamide with 0.1% bovine serum albumin/0.1% Ficoll/0.1% polyvinylpyrrolidone/50 mM sodium phosphate buffer at pH 6.5 with 750 mM sodium chloride, 75 mM sodium citrate at 42 C; or (3) overnight hybridization in a solution that employs 50% formamide, 5×SSC (0.75 M NaCl, 0.075 M sodium citrate), 50 mM sodium phosphate (pH 6.8), 0.1% sodium pyrophosphate, 5×Denhardt's solution, sonicated salmon sperm DNA (50 g/ml), 0.1% SDS, and 10% dextran sulfate at 42 C, with a 10 minute wash at 42 C in 0.2×SSC (sodium chloride/sodium citrate) followed by a 10 minute high-stringency wash consisting of 0.1×SSC containing EDTA at 55 C.
[0131] "Moderately stringent conditions" can be identified as described by Sambrook et al., Molecular Cloning: A Laboratory Manual, New York: Cold Spring Harbor Press, 1989, and include the use of washing solution and hybridization conditions (e.g., temperature, ionic strength and % SDS) less stringent that those described above. An example of moderately stringent conditions is overnight incubation at 37° C. in a solution comprising: 20% formamide, 5×SSC (150 mM NaCl, 15 mM trisodium citrate), 50 mM sodium phosphate (pH 7.6), 5×Denhardt's solution, 10% dextran sulfate, and 20 mg/ml denatured sheared salmon sperm DNA, followed by washing the filters in 1×SSC at about 37-50 C. The skilled artisan will recognize how to adjust the temperature, ionic strength, etc. as necessary to accommodate factors such as probe length and the like.
[0132] The term "epitope tagged" when used herein refers to a chimeric polypeptide comprising a STOP-1 polypeptide or anti-STOP-1 antibody fused to a "tag polypeptide". The tag polypeptide has enough residues to provide an epitope against which an antibody can be made, yet is short enough such that it does not interfere with activity of the polypeptide to which it is fused. The tag polypeptide preferably also is fairly unique so that the antibody does not substantially cross-react with other epitopes. Suitable tag polypeptides generally have at least six amino acid residues and usually between about 8 and 50 amino acid residues (preferably, between about 10 and 20 amino acid residues). Polypeptides and antibodies of this invention that are epitope-tagged are contemplated.
[0133] "Biologically active" and "biological activity" and "biological characteristics" with respect to an STOP-1 means (1) having the ability to increase cell proliferation of at least one type of mammalian cell (e.g., 3T3) in vivo or ex vivo; (2) having the ability to specifically bind STOP-1; and/or (3) having the ability to otherwise modulate STOP-1 signaling or STOP-1 activity, except where specified otherwise.
[0134] "Biologically active" and "biological activity" and "biological characteristics" with respect to a modified STOP-1 polypeptide or a STOP-1 polypeptide (1) having the ability to partially or fully block, inhibit or neutralize a biological activity of a native STOP-1 (either in an antagonistic or blocking manner); (2) having the ability to specifically bind STOP-1; and/or (3) having the ability to modulate STOP-1 signaling or STOP-1 activity, except where specified otherwise.
[0135] "Biologically active" and "biological activity" and "biological characteristics" with respect to an anti-STOP-1 antibody of this invention means (1) having the ability to partially or fully block, inhibit or neutralize a biological activity of a native STOP-1 (either in an antagonistic or blocking manner); (2) having the ability to specifically bind STOP-1; and/or (3) having the ability to modulate STOP-1 signaling or STOP-1 activity, except where specified otherwise. In one preferred embodiment, an antibody of this invention binds to STOP-1 with an affinity of at least 1 uM or less, 100 nm or less, 50 nm or less, 10 nm or less, 5 nM or less, 1 nm or less. As used herein, "antibody variable domain" refers to the portions of the light and heavy chains of antibody molecules that include amino acid sequences of Complementary Determining Regions (CDRs; ie., CDR1, CDR2, and CDR3), and Framework Regions (FRs). VH refers to the variable domain of the heavy chain. VL refers to the variable domain of the light chain. According to the methods used in this invention, the amino acid positions assigned to CDRs and FRs are defined according to Kabat (Sequences of Proteins of Immunological Interest (National Institutes of Health, Bethesda, Md., 1987 and 1991)) Amino acid numbering of antibodies or antigen binding fragments is also according to that of Kabat.
[0136] As used herein, "codon set" refers to a set of different nucleotide triplet sequences used to encode desired variant amino acids. A set of oligonucleotides can be synthesized, for example, by solid phase synthesis, containing sequences that represent all possible combinations of nucleotide triplets provided by the codon set and that will encode the desired group of amino acids. A standard form of codon designation is that of the IUB code, which is known in the art and described herein. A "non-random codon set", as used herein, thus refers to a codon set that encodes select amino acids that fulfill partially, preferably completely, the criteria for amino acid selection as described herein. Synthesis of oligonucleotides with selected nucleotide "degeneracy" at certain positions is known in that art, for example the TRIM approach (Knappek et al.; J. Mol. Biol. (1999), 296:57-86); Garrard & Henner, Gene (1993), 128:103). Such sets of nucleotides having certain codon sets can be synthesized using commercial nucleic acid synthesizers (available from, for example, Applied Biosystems, Foster City, Calif.), or can be obtained commercially (for example, from Life Technologies, Rockville, Md.). Therefore, a set of oligonucleotides synthesized having a particular codon set will typically include a plurality of oligonucleotides with different sequences, the differences established by the codon set within the overall sequence. Oligonucleotides, as used according to the invention, have sequences that allow for hybridization to a variable domain nucleic acid template and also can, but does not necessarily, include restriction enzyme sites useful for, for example, cloning purposes.
[0137] "Heterologous DNA" is any DNA that is introduced into a host cell. The DNA can be derived from a variety of sources including genomic DNA, cDNA, synthetic DNA and fusions or combinations of these. The DNA can include DNA from the same cell or cell type as the host or recipient cell or DNA from a different cell type, for example, from a mammal or plant. The DNA can, optionally, include marker or selection genes, for example, antibiotic resistance genes, temperature resistance genes, etc. Host cells encoding heterologous DNAs comprising the UNQ polypeptides and antibodies of this invention are contemplated as well as their use.
[0138] As used herein, "highly diverse position" refers to a position of an amino acid located in the variable regions of the light and heavy chains that have a number of different amino acid represented at the position when the amino acid sequences of known and/or naturally occurring antibodies or antigen binding fragments are compared. The highly diverse positions are typically in the CDR regions. In one aspect, the ability to determine highly diverse positions in known and/or naturally occurring antibodies is facilitated by the data provided by Kabat, Sequences of Proteins of Immunological Interest (National Institutes of Health, Bethesda, Md., 1987 and 1991). An internet-based database located at http://immuno bme.nwu.edu provides an extensive collection and alignment of human light and heavy chain sequences and facilitates determination of highly diverse positions in these sequences. According to the invention, an amino acid position is highly diverse if it has preferably from about 2 to about 11, preferably from about 4 to about 9, and preferably from about 5 to about 7 different possible amino acid residue variations at that position. In some embodiments, an amino acid position is highly diverse if it has preferably at least about 2, preferably at least about 4, preferably at least about 6, and preferably at least about 8 different possible amino acid residue variations at that position.
[0139] As used herein, "library" refers to a plurality of antibody or antibody fragment sequences (for example, polypeptides of the invention), or the nucleic acids that encode these sequences, the sequences being different in the combination of variant amino acids that are introduced into these sequences according to the methods of the invention.
[0140] "Phage display" is a technique by which variant polypeptides are displayed as fusion proteins to a coat protein on the surface of phage, e.g., filamentous phage, particles. A utility of phage display lies in the fact that large libraries of randomized protein variants can be rapidly and efficiently sorted for those sequences that bind to a target molecule with high affinity. Display of peptide and protein libraries on phage has been used for screening millions of polypeptides for ones with specific binding properties. Polyvalent phage display methods have been used for displaying small random peptides and small proteins through fusions to either gene III or gene VIII of filamentous phage. Wells and Lowman, Curr. Opin. Struct. Biol., 3:355-362 (1992), and references cited therein. In monovalent phage display, a protein or peptide library is fused to a gene III or a portion thereof, and expressed at low levels in the presence of wild type gene III protein so that phage particles display one copy or none of the fusion proteins. Avidity effects are reduced relative to polyvalent phage so that sorting is on the basis of intrinsic ligand affinity, and phagemid vectors are used, which simplify DNA manipulations. Lowman and Wells, Methods: A companion to Methods in Enzymology, 3:205-0216 (1991).
[0141] A "phagemid" is a plasmid vector having a bacterial origin of replication, e.g., Co1E1, and a copy of an intergenic region of a bacteriophage. The phagemid can be used on any known bacteriophage, including filamentous bacteriophage and lambdoid bacteriophage. The plasmid will also generally contain a selectable marker for antibiotic resistance. Segments of DNA cloned into these vectors can be propagated as plasmids. When cells harboring these vectors are provided with all genes necessary for the production of phage particles, the mode of replication of the plasmid changes to rolling circle replication to generate copies of one strand of the plasmid DNA and package phage particles. The phagemid can form infectious or non-infectious phage particles. This term includes phagemids which contain a phage coat protein gene or fragment thereof linked to a heterologous polypeptide gene as a gene fusion such that the heterologous polypeptide is displayed on the surface of the phage particle.
[0142] The term "phage vector" means a double stranded replicative form of a bacteriophage containing a heterologous gene and capable of replication. The phage vector has a phage origin of replication allowing phage replication and phage particle formation. The phage is preferably a filamentous bacteriophage, such as an M13, f1, fd, Pf3 phage or a derivative thereof, or a lambdoid phage, such as lambda, 21, phi80, phi81, 82, 424, 434, etc., or a derivative thereof.
[0143] As used herein, "target amino acid" refers to an amino acid that belongs to the group of amino acids that are collectively the most commonly occurring amino acids found at a particular position of known and/or natural antibodies or antigen binding fragments. In some embodiments, the most commonly occurring amino acids" are those amino acids that are found in a particular position in preferably at least about 50%, preferably at least about 70%, preferably at least about 80%, preferably at least about 90%, preferably all of sequences of known and/or natural antibodies or antigen binding fragments. In some embodiments, the most commonly occurring amino acids" are those amino acids that are found in a particular position in preferably from about 50% to about 100%, preferably from about 60% to about 90%, preferably from about 70% to about 85%, preferably from about 80% to about 85% of the sequences of known and/or natural antibodies or antigen binding fragments. Known antibodies or antigen biding fragments are those whose sequences are available in the art, such as those available in publicly-accessible databases, such as the database of Kabat ("Sequence of Proteins of Immunological Interest, National Institutes of Health, Bethesda, Md., 1987 and 1991) and/or as located at http://immuno.bme.nwu.edu. The amino acid position is preferably a position in the CDR region. A target group of amino acids refers to a group of target amino acids for a particular position. Preferably, a target amino acid is not a cysteine residue. For positions in the light chain CDR1, CDR2, CDR3, and for heavy chain CDR1 and CDR2, typically, a target group of amino acids can include from preferably about two to about eleven, preferably from about 4 to about 9, preferably from about 5 to about 7, preferably about 6 amino acids at a particular highly diverse and solvent-accessible position of the source sequence.
[0144] The term "proteoglycan" refers to a molecule where at least one glycosaminoglycan side chain is covalently attached to the protein core of the molecule. A proteoglycan synthesis deficient cell line according to this invention includes a cell line that is deficient in galactosyltransferase I. According to one preferred embodiment, the cell line is a CHO-psbg cell line.
[0145] The term "antagonist" is any molecule that partially or fully blocks, inhibits, or neutralizes a biological activity of a native STOP-1 polypeptide and that specifically binds to a native STOP-1 polypeptide, wherein the binding of the antagonist (1) is to a native STOP-1 polypeptide in oligomeric form, (2) is to residues 94-243 of native human STOP-1 and/or (3) can be inhibited (e.g., as observed in a competitive ELISA assay using STOP-1 and 6B12) by a monoclonal antibody of this invention (e.g., a deposited antibody of this invention, etc.). According to one embodiment, the antagonist is a polypeptide. According to another embodiment, the 6B12 antibody can inhibit the binding of the antagonist to STOP-1. According to another embodiment, the antagonist binds to a residue within residues 33-52 or 33-53 of human STOP-1 or a non-human STOP-1 equivalent thereof.
[0146] The term "small molecule antagonist" refers to any molecule wherein the molecular weight is 1500 daltons or less and is an antagonist according to this invention. According to one embodiment the small molecule antagonist is below about 500 Daltons.
[0147] According to one preferred embodiment, the antagonist blocks, inhibits or neutralizes cell proliferation in cells expressing native STOP-1. In one embodiment, the antagonist or small molecule antagonist prevents migration of a cell to which STOP-1 binds. In one preferred embodiment, the antagonist or small molecule antagonist specifically binds to a trimeric form of STOP-1. Suitable antagonists include antibodies, amino acid sequence variants of native STOP-1 polypeptides, peptides, of this invention etc. Methods for identifying antagonists of a STOP-1 polypeptide can comprise contacting a STOP-1 polypeptide with a candidate antagonist molecule and measuring a detectable change in one or more biological activities associated with the STOP-1 polypeptide.
[0148] The term "potentiator" refers to any molecule that enhances a biological activity of a native STOP-1 polypeptide and that specifically binds to a native STOP-1 polypeptide, wherein the potentiator can bind to an oligomeric form of a native STOP-1 polypeptide and has at least one additional activity selected from the group consisting of (1) is capable of binding to a residue in residues 94-243 of native human STOP-1, (2) is capable of aggregating STOP-1 on a cell; and (3) can be competed by a monoclonal antibody an S7 or S4 antibody of this invention (e.g., as observed in a competitive ELISA assay using STOP-1, S7 or S4 and the potentiator). According to one embodiment, the potentiator increases the binding of STOP-1 to a cell (e.g., HUVEC cells, HeLa cells, and HT1080 cells). In one preferred embodiment, the agonist binds to a trimeric form of STOP-1. Methods for identifying agonists of a STOP-1 polypeptide can comprise contacting a cell that binds STOP-1 with a STOP-1 polypeptide and the candidate agonist and measuring a detectable change in one or more biological activities associated with the STOP-1 polypeptide (e.g., increased binding of the STOP-1 polypeptide, cell proliferation or cell migration).
[0149] "Treating" or "treatment" or "alleviation" refers to both therapeutic treatment and prophylactic or preventative measures, wherein the object is to prevent or slow down (lessen) the targeted pathologic condition or disorder. Those in need of treatment include those already with the disorder as well as those prone to have the disorder or those in whom the disorder is to be prevented. These terms indicate the therapeutic and prophylactic uses herein are successful if they ameliorate, lessen or decrease the symptoms, complications or other problems associated with a disease or ameliorate, lessen or decrease the chance of onset or frequency of the symptoms, complications or other problems associated with a disease.
[0150] A subject or mammal is successfully "treated" for a STOP-1 polypeptide-expressing cancer if, after receiving a therapeutic amount of an antagonist according to the methods of the present invention, the patient shows observable and/or measurable reduction in or absence of one or more of the following: reduction in the number of cancer cells or absence of the cancer cells; reduction in the tumor size; inhibition (i.e., slow to some extent and preferably stop) of cancer cell infiltration into peripheral organs including the spread of cancer into soft tissue and bone; inhibition (i.e., slow to some extent and preferably stop) of tumor metastasis; inhibition, to some extent, of tumor growth; and/or relief to some extent, one or more of the symptoms associated with the specific cancer; reduced morbidity and mortality, and improvement in quality of life issues. To the extent the anti-STOP-1 antibody or STOP-1 binding oligopeptide can prevent growth and/or kill existing cancer cells, it can be cytostatic and/or cytotoxic. Reduction of these signs or symptoms can also be felt by the patient.
[0151] A subject or mammal is successfully "treated" for abnormal angiogenesis if, after receiving a therapeutic amount of an antagonist or agonist according to the methods of the present invention, the patient shows observable and/or measurable [TO BE FILLED IN]; and/or relief to some extent, one or more of the symptoms associated with the abnormal angiogenesis; and improvement in quality of life issues.
[0152] The above parameters for assessing successful treatment and improvement in the disease are readily measurable by procedures familiar to a physician. For cancer therapy, efficacy can be measured, for example, by assessing the time to disease progression (TTP) and/or determining the response rate (RR). Metastasis can be determined by staging tests and by bone scan and tests for calcium level and other enzymes to determine spread to the bone. CT scans can also be done to look for spread to the pelvis and lymph nodes in the area. Chest X-rays and measurement of liver enzyme levels by known methods are used to look for metastasis to the lungs and liver, respectively. Other known methods for monitoring the disease include transrectal ultrasonography (TRUS) and transrectal needle biopsy (TRNB).
[0153] For bladder cancer, which is a more localized cancer, methods to determine progress of disease include urinary cytologic evaluation by cystoscopy, monitoring for presence of blood in the urine, visualization of the urothelial tract by sonography or an intravenous pyelogram, computed tomography (CT) and magnetic resonance imaging (MRI). The presence of distant metastases can be assessed by CT of the abdomen, chest x-rays, or radionuclide imaging of the skeleton.
[0154] "Chronic" administration refers to administration of the agent(s) in a continuous mode as opposed to an acute mode, so as to maintain the initial therapeutic effect (activity) for an extended period of time. "Intermittent" administration is treatment that is not consecutively done without interruption, but rather is cyclic in nature.
[0155] "Mammal" for purposes of the treatment of, alleviating the symptoms of or diagnosis of a cancer refers to any animal classified as a mammal (aka "patient"), including humans, domestic and farm animals, and zoo, sports, or pet animals, such as dogs, cats, cattle, horses, sheep, pigs, goats, rabbits, etc. Preferably, the mammal is human.
[0156] Administration "in combination with" one or more further therapeutic agents includes simultaneous (concurrent) and consecutive administration in any order.
[0157] "Carriers" as used herein include pharmaceutically acceptable carriers, excipients, or stabilizers which are nontoxic to the cell or mammal being exposed thereto at the dosages and concentrations employed. Often the physiologically acceptable carrier is an aqueous pH buffered solution. Examples of physiologically acceptable carriers include buffers such as phosphate, citrate, and other organic acids; antioxidants including ascorbic acid; low molecular weight (less than about 10 residues) polypeptide; proteins, such as serum albumin, gelatin, or immunoglobulins; hydrophilic polymers such as polyvinylpyrrolidone; amino acids such as glycine, glutamine, asparagine, arginine or lysine; monosaccharides, disaccharides, and other carbohydrates including glucose, mannose, or dextrins; chelating agents such as EDTA; sugar alcohols such as mannitol or sorbitol; salt-forming counterions such as sodium; and/or nonionic surfactants such as TWEEN®, polyethylene glycol (PEG), and PLURONICS®.
[0158] By "solid phase" or "solid support" is meant a non-aqueous matrix to which an antibody, an antagonist or a polypeptide of the present invention can adhere or attach. Examples of solid phases encompassed herein include those formed partially or entirely of glass (e.g., controlled pore glass), polysaccharides (e.g., agarose), polyacrylamides, polystyrene, polyvinyl alcohol and silicones. In certain embodiments, depending on the context, the solid phase can comprise the well of an assay plate; in others it is a purification column (e.g., an affinity chromatography column). This term also includes a discontinuous solid phase of discrete particles, such as those described in U.S. Pat. No. 4,275,149.
[0159] As used herein, the term "immunoadhesin" designates antibody-like molecules that combine the binding specificity of a heterologous protein (an "adhesin") with the effector functions of immunoglobulin constant domains Structurally, the immunoadhesins comprise a fusion of an amino acid sequence with the desired binding specificity that is other than the antigen recognition and binding site of an antibody (i.e., is "heterologous"), and an immunoglobulin constant domain sequence. The adhesin part of an immunoadhesin molecule typically is a contiguous amino acid sequence comprising at least the binding site of a receptor or a ligand--such as a portion of a native STOP-1 protein. The immunoglobulin constant domain sequence in the immunoadhesin can be obtained from any immunoglobulin, such as IgG-1, IgG-2, IgG-3, or IgG-4 subtypes, IgA (including IgA-1 and IgA-2), IgE, IgD, or IgM.
[0160] A "liposome" is a small vesicle composed of various types of lipids, phospholipids and/or surfactant which is useful for delivery of a drug (such as a STOP-1 polypeptide, an antibody thereto or a STOP-1 binding oligopeptide) to a mammal. The components of the liposome are commonly arranged in a bilayer formation, similar to the lipid arrangement of biological membranes.
[0161] A "small" molecule or "small" organic molecule is defined herein to have a molecular weight below about 500 Daltons.
[0162] An "effective amount" of a polypeptide, antibody, antagonist or composition as disclosed herein is an amount sufficient to carry out a specifically stated purpose. An "effective amount" can be determined empirically and by known methods relating to the stated purpose.
[0163] The term "therapeutically effective amount" refers to an amount of an antibody, polypeptide or antagonist of this invention effective to "treat" a disease or disorder in a mammal (aka patient). In the case of cancer, the therapeutically effective amount of the drug can reduce the number of cancer cells; reduce the tumor size; inhibit (i.e., slow to some extent and preferably stop) cancer cell infiltration into peripheral organs; inhibit (i.e., slow to some extent and preferably stop) tumor metastasis; inhibit, to some extent, tumor growth; and/or relieve to some extent one or more of the symptoms associated with the cancer. See the definition herein of "treating". To the extent the drug can prevent growth and/or kill existing cancer cells, it can be cytostatic and/or cytotoxic.
[0164] A "growth inhibitory amount" of a polypeptide, antibody, antagonist or composition of this invention is an amount capable of inhibiting the growth of a cell, especially tumor, e.g., cancer cell, either in vitro or in vivo. A "growth inhibitory amount" of a polypeptide, antibody, antagonist or composition of this invention for purposes of inhibiting neoplastic cell growth can be determined empirically and by known methods or by examples provided herein.
[0165] A "cytotoxic amount" of a polypeptide, antibody, antagonist or composition of this invention is an amount capable of causing the destruction of a cell, especially tumor, e.g., cancer cell, either in vitro or in vivo. A "cytotoxic amount" of a polypeptide, antibody, antagonist or composition of this invention for purposes of inhibiting neoplastic cell growth can be determined empirically and by methods known in the art.
[0166] The term "antibody" is used in the broadest sense and specifically covers, for example, single anti-STOP-1 monoclonal antibodies (including agonist, antagonist, and neutralizing antibodies), anti-STOP-1 antibody compositions with polyepitopic specificity, polyclonal antibodies, single chain anti-STOP-1 antibodies, and fragments of anti-STOP-1 antibodies (see below) as long as they specifically bind a native STOP-1 polypeptide and/or exhibit a biological activity or immunological activity of this invention. According to one embodiment, the antibody binds to an oligomeric form of STOP-1, e.g., a trimeric form. In a further embodiment, the antibody specifically binds to human STOP-1 between residues 94-243. According to another embodiment, the antibody specifically binds to STOP-1, which binding can be inhibited by a monoclonal antibody of this invention (e.g., a deposited antibody of this invention, etc.). The phrase "functional fragment or analog" of an antibody is a compound having a qualitative biological activity in common with an antibody to which it is being referred. For example, a functional fragment or analog of an anti-STOP-1 antibody can be one which can specifically bind to a STOP-1 molecule. In one embodiment, the antibody can prevent or substantially reduce the ability of a STOP-1 molecule to induce cell proliferation. The term "immunoglobulin" (Ig) is used interchangeably with "antibody" herein. According to one embodiment, an antibody of this invention does not bind to a peptide having the amino acid sequence GWNSVSRIIIEELPK (SEQ ID NO:117).
[0167] An "isolated antibody" is one which has been identified and separated and/or recovered from a component of its natural environment. Contaminant components of its natural environment are materials which would interfere with diagnostic or therapeutic uses for the antibody, and can include enzymes, hormones, and other proteinaceous or nonproteinaceous solutes. In preferred embodiments, the antibody will be purified (1) to greater than 95% by weight of antibody as determined by the Lowry method, and most preferably more than 99% by weight, (2) to a degree sufficient to obtain at least 15 residues of N-terminal or internal amino acid sequence by use of a spinning cup sequenator, or (3) to homogeneity by SDS-PAGE under reducing or nonreducing conditions using Coomassie blue or, preferably, silver stain. Isolated antibody includes the antibody in situ within recombinant cells since at least one component of the antibody's natural environment will not be present. Ordinarily, however, isolated antibody will be prepared by at least one purification step.
[0168] The basic 4-chain antibody unit is a heterotetrameric glycoprotein composed of two identical light (L) chains and two identical heavy (H) chains (an IgM antibody consists of 5 of the basic heterotetramer unit along with an additional polypeptide called J chain, and therefore contain 10 antigen binding sites, while secreted IgA antibodies can polymerize to form polyvalent assemblages comprising 2-5 of the basic 4-chain units along with J chain) In the case of IgGs, the 4-chain unit is generally about 150,000 daltons. Each L chain is linked to a H chain by one covalent disulfide bond, while the two H chains are linked to each other by one or more disulfide bonds depending on the H chain isotype. Each H and L chain also has regularly spaced intrachain disulfide bridges. Each H chain has at the N-terminus, a variable domain (VH) followed by three constant domains (CH) for each of the and chains and four CH domains for and isotypes. Each L chain has at the N-terminus, a variable domain (VL) followed by a constant domain (CL) at its other end. The VL is aligned with the VH and the CL is aligned with the first constant domain of the heavy chain (CH1). Particular amino acid residues are believed to form an interface between the light chain and heavy chain variable domains. The pairing of a VH and VL together forms a single antigen-binding site. For the structure and properties of the different classes of antibodies, see, e.g., Basic and Clinical Immunology, 8th edition, Daniel P. Stites, Abba I. Terr and Tristram G. Parslow (eds.), Appleton & Lange, Norwalk, Conn., 1994, page 71 and Chapter 6.
[0169] The L chain from any vertebrate species can be assigned to one of two clearly distinct types, called kappa and lambda, based on the amino acid sequences of their constant domains Depending on the amino acid sequence of the constant domain of their heavy chains (CH), immunoglobulins can be assigned to different classes or isotypes. There are five classes of immunoglobulins: IgA, IgD, IgE, IgG, and IgM, having heavy chains designated, , , , and, respectively. The and classes are further divided into subclasses on the basis of relatively minor differences in CH sequence and function, e.g., humans express the following subclasses: IgG1, IgG2, IgG3, IgG4, IgA1, and IgA2.
[0170] The term "variable" refers to the fact that certain segments of the variable domains differ extensively in sequence among antibodies. The V domain mediates antigen binding and define specificity of a particular antibody for its particular antigen. However, the variability is not evenly distributed across the 110-amino acid span of the variable domains Instead, the V regions consist of relatively invariant stretches called framework regions (FRs) of 15-30 amino acids separated by shorter regions of extreme variability called "hypervariable regions" that are each 9-12 amino acids long. The variable domains of native heavy and light chains each comprise four FRs, largely adopting a sheet configuration, connected by three hypervariable regions, which form loops connecting, and in some cases forming part of, the sheet structure. The hypervariable regions in each chain are held together in close proximity by the FRs and, with the hypervariable regions from the other chain, contribute to the formation of the antigen-binding site of antibodies (see Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, Md. (1991)). The constant domains are not involved directly in binding an antibody to an antigen, but exhibit various effector functions, such as participation of the antibody in antibody dependent cellular cytotoxicity (ADCC).
[0171] The term "hypervariable region" when used herein refers to the amino acid residues of an antibody which are responsible for antigen-binding. The hypervariable region generally comprises amino acid residues from a "complementarity determining region" or "CDR" (e.g. around about residues 24-34 (L1), 50-56 (L2) and 89-97 (L3) in the VL, and around about 1-35 (H1), 50-65 (H2) and 95-102 (H3) in the VH (in one embodiment, H1 is around about 31-35); Kabat et al., Sequences of Proteins of Immunological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, Md. (1991)) and/or those residues from a "hypervariable loop" (e.g. residues 26-32 (L1), 50-52 (L2) and 91-96 (L3) in the VL, and 26-32 (H1), 53-55 (H2) and 96-101 (H3) in the VH; Chothia and Lesk J. Mol. Biol. 196:901-917 (1987)).
[0172] The term "monoclonal antibody" as used herein refers to an antibody obtained from a population of substantially homogeneous antibodies, i.e., the individual antibodies comprising the population are identical except for possible naturally occurring mutations that can be present in minor amounts. Monoclonal antibodies are highly specific, being directed against a single antigenic site. Furthermore, in contrast to polyclonal antibody preparations which include different antibodies directed against different determinants (epitopes), each monoclonal antibody is directed against a single determinant on the antigen. In addition to their specificity, the monoclonal antibodies are advantageous in that they can be synthesized uncontaminated by other antibodies. The modifier "monoclonal" is not to be construed as requiring production of the antibody by any particular method. For example, the monoclonal antibodies useful in the present invention can be prepared by the hybridoma methodology first described by Kohler et al., Nature, 256:495 (1975), or can be made using recombinant DNA methods in bacterial, eukaryotic animal or plant cells (see, e.g., U.S. Pat. No. 4,816,567). The "monoclonal antibodies" can also be isolated from phage antibody libraries using the techniques described in Clackson et al., Nature, 352:624-628 (1991), Marks et al., J. Mol. Biol., 222:581-597 (1991), and the Examples below, for example.
[0173] The monoclonal antibodies herein include "chimeric" antibodies in which a portion of the heavy and/or light chain is identical with or homologous to corresponding sequences in antibodies derived from a particular species or belonging to a particular antibody class or subclass, while the remainder of the chain(s) is identical with or homologous to corresponding sequences in antibodies derived from another species or belonging to another antibody class or subclass, as well as fragments of such antibodies, so long as they exhibit a biological activity of this invention (see U.S. Pat. No. 4,816,567; and Morrison et al., Proc. Natl. Acad. Sci. USA, 81:6851-6855 (1984)). Chimeric antibodies of interest herein include "primatized" antibodies comprising variable domain antigen-binding sequences derived from a non-human primate (e.g. Old World Monkey, Ape etc), and human constant region sequences.
[0174] An "intact" antibody is one which comprises an antigen-binding site as well as a CL and at least heavy chain constant domains, CH1, CH2 and CH3. The constant domains can be native sequence constant domains (e.g. human native sequence constant domains) or amino acid sequence variant thereof. Preferably, the intact antibody has one or more effector functions.
[0175] "Antibody fragments" comprise a portion of an intact antibody, preferably the antigen binding or variable region of the intact antibody. Examples of antibody fragments include Fab, Fab', F(ab')2, and Fv fragments; diabodies; linear antibodies (see U.S. Pat. No. 5,641,870, Example 2; Zapata et al., Protein Eng. 8(10): 1057-1062 [1995]); single-chain antibody molecules; and multispecific antibodies formed from antibody fragments. The expression "linear antibodies" generally refers to the antibodies described in Zapata et al., Protein Eng., 8(10):1057-1062 (1995). Briefly, these antibodies comprise a pair of tandem Fd segments (VH-CH1-VH-CH1) which, together with complementary light chain polypeptides, form a pair of antigen binding regions. Linear antibodies can be bispecific or monospecific.
[0176] Papain digestion of antibodies produces two identical antigen-binding fragments, called "Fab" fragments, and a residual "Fc" fragment, a designation reflecting the ability to crystallize readily. The Fab fragment consists of an entire L chain along with the variable region domain of the H chain (VH), and the first constant domain of one heavy chain (CH1). Each Fab fragment is monovalent with respect to antigen binding, i.e., it has a single antigen-binding site. Pepsin treatment of an antibody yields a single large F(ab')2 fragment which roughly corresponds to two disulfide linked Fab fragments having divalent antigen-binding activity and is still capable of cross-linking antigen. Fab' fragments differ from Fab fragments by having additional few residues at the carboxy terminus of the CH1 domain including one or more cysteines from the antibody hinge region. Fab'-SH is the designation herein for Fab' in which the cysteine residue(s) of the constant domains bear a free thiol group. F(ab')2 antibody fragments originally were produced as pairs of Fab' fragments which have hinge cysteines between them. Other chemical couplings of antibody fragments are also known.
[0177] The Fc fragment comprises the carboxy-terminal portions of both H chains held together by disulfides. The effector functions of antibodies are determined by sequences in the Fc region, which region is also the part recognized by Fc receptors (FcR) found on certain types of cells.
[0178] "Fv" is the minimum antibody fragment which contains a complete antigen-recognition and -binding site. This fragment consists of a dimer of one heavy- and one light-chain variable region domain in tight, non-covalent association. From the folding of these two domains emanate six hypervariable loops (3 loops each from the H and L chain) that contribute the amino acid residues for antigen binding and confer antigen binding specificity to the antibody. However, even a single variable domain (or half of an Fv comprising only three CDRs specific for an antigen) has the ability to recognize and bind antigen, although at a lower affinity than the entire binding site.
[0179] "Single-chain Fv" also abbreviated as "sFv" or "scFv" are antibody fragments that comprise the VH and VL antibody domains connected into a single polypeptide chain. Preferably, the sFv polypeptide further comprises a polypeptide linker between the VH and VL domains which enables the sFv to form the desired structure for antigen binding. For a review of sFv, see Pluckthun in The Pharmacology of Monoclonal Antibodies, vol. 113, Rosenburg and Moore eds., Springer-Verlag, New York, pp. 269-315 (1994); Borrebaeck 1995, infra.
[0180] The term "diabodies" refers to small antibody fragments prepared by constructing sFv fragments (see preceding paragraph) with short linkers (about 5-10 residues) between the VH and VL domains such that inter-chain but not intra-chain pairing of the V domains is achieved, resulting in a bivalent fragment, i.e., fragment having two antigen-binding sites. Bispecific diabodies are heterodimers of two "crossover" sFv fragments in which the VH and VL domains of the two antibodies are present on different polypeptide chains. Diabodies are described more fully in, for example, EP 404,097; WO 93/11161; and Hollinger et al., Proc. Natl. Acad. Sci. USA, 90:6444-6448 (1993).
[0181] "Humanized" forms of non-human (e.g., rodent) antibodies are chimeric antibodies that contain minimal sequence derived from the non-human antibody. For the most part, humanized antibodies are human immunoglobulins (recipient antibody) in which residues from a hypervariable region of the recipient are replaced by residues from a hypervariable region of a non-human species (donor antibody) such as mouse, rat, rabbit or non-human primate having the desired antibody specificity, affinity, and capability. In some instances, framework region (FR) residues of the human immunoglobulin are replaced by corresponding non-human residues. Furthermore, humanized antibodies can comprise residues that are not found in the recipient antibody or in the donor antibody. These modifications are made to further refine antibody performance. In general, the humanized antibody will comprise substantially all of at least one, and typically two, variable domains, in which all or substantially all of the hypervariable loops correspond to those of a non-human immunoglobulin and all or substantially all of the FRs are those of a human immunoglobulin sequence. The humanized antibody optionally also will comprise at least a portion of an immunoglobulin constant region (Fc), typically that of a human immunoglobulin. For further details, see Jones et al., Nature 321:522-525 (1986); Riechmann et al., Nature 332:323-329 (1988); and Presta, Curr. Op. Struct. Biol. 2:593-596 (1992).
[0182] A "species-dependent antibody," e.g., a mammalian anti-human IgE antibody, is an antibody which has a stronger binding affinity for an antigen from a first mammalian species than it has for a homologue of that antigen from a second mammalian species. Normally, the species-dependent antibody "bind specifically" to a human antigen (i.e., has a binding affinity (Kd) value of no more than about 1×10-7 M, preferably no more than about 1×10-8 and most preferably no more than about 1×10-9 M) but has a binding affinity for a homologue of the antigen from a second non-human mammalian species which is at least about 50 fold, or at least about 500 fold, or at least about 1000 fold, weaker than its binding affinity for the human antigen. The species-dependent antibody can be of any of the various types of antibodies as defined above, but preferably is a humanized or human antibody.
[0183] A "STOP-1 binding oligopeptide" is an oligopeptide that binds, preferably specifically, to a STOP-1 polypeptide as described herein. STOP-1 binding oligopeptides can be chemically synthesized using known oligopeptide synthesis methodology or can be prepared and purified using recombinant technology. STOP-1 binding oligopeptides are usually at least about 5 amino acids in length, alternatively at least about 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, or 100 amino acids in length or more, wherein such oligopeptides that are capable of binding, preferably specifically, to a STOP-1 polypeptide as described herein. According to one embodiment, the STOP-1 binding oligopeptide binds to the same or overlapping region that the 6B12 antibody binds. STOP-1 binding oligopeptides can be identified without undue experimentation using known techniques. In this regard, it is noted that techniques for screening oligopeptide libraries for oligopeptides that are capable of specifically binding to a polypeptide target are known in the art (see, e.g., U.S. Pat. Nos. 5,556,762, 5,750,373, 4,708,871, 4,833,092, 5,223,409, 5,403,484, 5,571,689, 5,663,143; PCT Publication Nos. WO 84/03506 and WO84/03564; Geysen et al., Proc. Natl. Acad. Sci. U.S.A., 81:3998-4002 (1984); Geysen et al., Proc. Natl. Acad. Sci. U.S.A., 82:178-182 (1985); Geysen et al., in Synthetic Peptides as Antigens, 130-149 (1986); Geysen et al., J. Immunol. Meth., 102:259-274 (1987); Schoofs et al., J. Immunol., 140:611-616 (1988), Cwirla, S. E. et al. (1990) Proc. Natl. Acad. Sci. USA, 87:6378; Lowman, H. B. et al. (1991) Biochemistry, 30:10832; Clackson, T. et al. (1991) Nature, 352: 624; Marks, J. D. et al. (1991), J. Mol. Biol., 222:581; Kang, A. S. et al. (1991) Proc. Natl. Acad. Sci. USA, 88:8363, and Smith, G. P. (1991) Current Opin. Biotechnol., 2:668).
[0184] A polypeptide, antibody, antagonist or composition of this invention "which binds" an antigen of interest, e.g. a tumor-associated polypeptide antigen target such as STOP-1, is one that binds the antigen with sufficient affinity such that a polypeptide, antibody, antagonist or composition is useful as a diagnostic and/or therapeutic agent in targeting a cell or tissue expressing the antigen, and does not significantly cross-react with other proteins. In such embodiments, the extent of binding of the polypeptide, antibody, antagonist or composition to a "non-target" protein will be less than about 10% of the binding of the polypeptide, antibody, antagonist or composition to its particular target protein as determined by fluorescence activated cell sorting (FACS) analysis or radioimmunoprecipitation (RIA). With regard to the binding of a polypeptide, antibody, antagonist or composition to a target molecule, the term "specific binding" or "specifically binds to" or is "specific for" a particular polypeptide or an epitope on a particular polypeptide target means binding that is measurably different from a non-specific interaction. Specific binding can be measured, for example, by determining binding of a molecule compared to binding of a control molecule, which generally is a molecule of similar structure that does not have binding activity. For example, specific binding can be determined by competition with a control molecule that is similar to the target, for example, an excess of non-labeled target. In this case, specific binding is indicated if the binding of the labeled target to a probe is competitively inhibited by excess unlabeled target. The term "specific binding" or "specifically binds to" or is "specific for" a particular polypeptide or an epitope on a particular polypeptide target as used herein can be exhibited, for example, by a molecule having a Kd for the target of at least about 10-4 M, alternatively at least about 10-5 M, alternatively at least about 10-6 M, alternatively at least about 10-7 M, alternatively at least about 10-8 M, alternatively at least about 10-9 M, alternatively at least about 10-10 M, alternatively at least about 10-11M, alternatively at least about 10-12 M, or greater. In one embodiment, the term "specific binding" refers to binding where a molecule binds to a particular polypeptide or epitope on a particular polypeptide without substantially binding to any other polypeptide or polypeptide epitope (e.g., a non-STOP-1 protein). It is understood that an antibody that specifically binds to a human native STOP-1 polypeptide may also bind a non-human native STOP-1 polypeptide.
[0185] A polypeptide, antibody, antagonist or composition that "inhibits the growth" of tumor cells expressing a STOP-1 polypeptide or a "growth inhibitory" polypeptide, antibody, antagonist or composition is one which results in measurable growth inhibition of cancer cells expressing or overexpressing the appropriate STOP-1 polypeptide. Preferred growth inhibitory polypeptides, antibodies, antagonists or compositions inhibit growth of STOP-1-expressing tumor cells by greater than 20%, preferably from about 20% to about 50%, and even more preferably, by greater than 50% (e.g., from about 50% to about 100%) as compared to the appropriate control, the control typically being tumor cells not treated with the polypeptide, antibody, antagonist or composition being tested. In one embodiment, growth inhibition can be measured at an antibody concentration of about 0.1 to 30 g/ml or about 0.5 nM to 200 nM in cell culture, where the growth inhibition is determined 1-10 days after exposure of the tumor cells to the antibody. Growth inhibition of tumor cells in vivo can be determined in various ways such as is described in the Experimental Examples section below. The antibody is growth inhibitory in vivo if administration of the anti-STOP-1 antibody at about 1 g/kg to about 100 mg/kg body weight results in reduction in tumor size or tumor cell proliferation within about 5 days to 3 months from the first administration of the antibody, preferably within about 5 to 30 days.
[0186] Antibody "effector functions" refer to those biological activities attributable to the Fc region (a native sequence Fc region or amino acid sequence variant Fc region) of an antibody, and vary with the antibody isotype. Examples of antibody effector functions include: C1q binding and complement dependent cytotoxicity; Fc receptor binding; antibody-dependent cell-mediated cytotoxicity (ADCC); phagocytosis; down regulation of cell surface receptors (e.g., B cell receptor); and B cell activation.
[0187] "Antibody-dependent cell-mediated cytotoxicity" or "ADCC" refers to a form of cytotoxicity in which secreted Ig bound onto Fc receptors (FcRs) present on certain cytotoxic cells (e.g., Natural Killer (NK) cells, neutrophils, and macrophages) enable these cytotoxic effector cells to bind specifically to an antigen-bearing target cell and subsequently kill the target cell with cytotoxins. The antibodies "arm" the cytotoxic cells and are absolutely required for such killing. The primary cells for mediating ADCC, NK cells, express Fc RIII only, whereas monocytes express Fc RI, Fc RII and Fc RIII. FcR expression on hematopoietic cells is summarized in Table 3 on page 464 of Ravetch and Kinet, Annu. Rev. Immunol. 9:457-92 (1991). To assess ADCC activity of a molecule of interest, an in vitro ADCC assay, such as that described in U.S. Pat. No. 5,500,362 or U.S. Pat. No. 5,821,337 can be performed. Useful effector cells for such assays include peripheral blood mononuclear cells (PBMC) and Natural Killer (NK) cells. Alternatively, or additionally, ADCC activity of the molecule of interest can be assessed in vivo, e.g., in a animal model such as that disclosed in Clynes et al. (USA) 95:652-656 (1998).
[0188] "Fc receptor" or "FcR" describes a receptor that binds to the Fc region of an antibody. The preferred FcR is a native sequence human FcR. Moreover, a preferred FcR is one which binds an IgG antibody (a gamma receptor) and includes receptors of the Fc RI, Fc RII and Fc RIII subclasses, including allelic variants and alternatively spliced forms of these receptors. Fc RII receptors include Fc RIIA (an "activating receptor") and Fc RIIB (an "inhibiting receptor"), which have similar amino acid sequences that differ primarily in the cytoplasmic domains thereof. Activating receptor Fc RIIA contains an immunoreceptor tyrosine-based activation motif (ITAM) in its cytoplasmic domain. Inhibiting receptor Fc RIIB contains an immunoreceptor tyrosine-based inhibition motif (ITIM) in its cytoplasmic domain. (see review M. in Daeron, Annu. Rev. Immunol 15:203-234 (1997)). FcRs are reviewed in Ravetch and Kinet, Annu. Rev. Immunol 9:457-492 (1991); Capel et al., Immunomethods 4:25-34 (1994); and de Haas et al., J. Lab. Clin. Med. 126:330-41 (1995). Other FcRs, including those to be identified in the future, are encompassed by the term "FcR" herein. The term also includes the neonatal receptor, FcRn, which is responsible for the transfer of maternal IgGs to the fetus (Guyer et al., J. Immunol. 117:587 (1976) and Kim et al., J. Immunol. 24:249 (1994)).
[0189] "Human effector cells" are leukocytes which express one or more FcRs and perform effector functions. Preferably, the cells express at least Fc RIII and perform ADCC effector function. Examples of human leukocytes which mediate ADCC include peripheral blood mononuclear cells (PBMC), natural killer (NK) cells, monocytes, cytotoxic T cells and neutrophils; with PBMCs and NK cells being preferred. The effector cells can be isolated from a native source, e.g., from blood.
[0190] "Complement dependent cytotoxicity" or "CDC" refers to the lysis of a target cell in the presence of complement. Activation of the classical complement pathway is initiated by the binding of the first component of the complement system (C1q) to antibodies (of the appropriate subclass) which are bound to their cognate antigen. To assess complement activation, a CDC assay, e.g., as described in Gazzano-Santoro et al., J. Immunol. Methods 202:163 (1996), can be performed.
[0191] The terms "cancer" and "cancerous" refer to or describe the physiological condition in mammals that is typically characterized by unregulated cell growth. Examples of cancer include, but are not limited to, carcinoma, lymphoma, blastoma, sarcoma, and leukemia or lymphoid malignancies. More particular examples of such cancers include squamous cell cancer (e.g., epithelial squamous cell cancer), lung cancer including small-cell lung cancer, non-small cell lung cancer, adenocarcinoma of the lung and squamous carcinoma of the lung, cancer of the peritoneum, hepatocellular cancer, gastric or stomach cancer including gastrointestinal cancer, pancreatic cancer, glioblastoma, cervical cancer, ovarian cancer, liver cancer, bladder cancer, cancer of the urinary tract, hepatoma, breast cancer, colon cancer, rectal cancer, colorectal cancer, endometrial or uterine carcinoma, salivary gland carcinoma, kidney or renal cancer, prostate cancer, vulval cancer, thyroid cancer, hepatic carcinoma, anal carcinoma, penile carcinoma, melanoma, multiple myeloma and B-cell lymphoma, brain, as well as head and neck cancer, and associated metastases.
[0192] The terms "cell proliferative disorder" and "proliferative disorder" refer to disorders that are associated with some degree of abnormal cell proliferation. In one embodiment, the cell proliferative disorder is cancer. In one embodiment, the cell proliferative disorder is desmoplasia.
[0193] "Tumor", as used herein, refers to all neoplastic cell growth and proliferation, whether malignant or benign, and all pre-cancerous and cancerous cells and tissues.
[0194] The term abnormal angiogenesis according to this invention occurs when new blood vessels either grow excessively, insufficiently or inappropriately (e.g., the location, timing or onset of the angiogenesis being undesired from a medical standpoint) in a diseased state or such that it causes a diseased state. "Excessive, inappropriate or uncontrolled angiogenesis" occurs in a diseased state when there is new blood vessel growth that contributes to the worsening of the diseased state, such as in cancer, especially vascularized solid tumors and metastatic tumores (including colon, lung cancer (especially small-cell lung cancer), or prostate cancer), diseases caused by ocular neovascularisation, especially diabetic blindness, retinopathies, primarily diabetic retinopathy or age-induced macular degeneration and rubeosis; psoriasis, haemangioblastoma such as haemangioma; inflammatory renal diseases, such as glomerulonephritis, especially mesangioproliferative glomerulonephritis, haemolytic uremic syndrome, diabetic nephropathy or hypertensive nephrosclerosis; various imflammatory diseases, such as arthritis, especially rheumatoid arthritis, inflammatory bowel disease, psorsasis, sarcoidosis, arterial arteriosclerosis and diseases occurring after transplants, endometriosis or chronic asthma and more than 70 other conditions. The new blood vessels can feed the diseased tissues, destroy normal tissues, and in the case of cancer, the new vessels can allow tumor cells to escape into the circulation and lodge in other organs (tumor metastases). Insufficient angiogenesis occurs when there is inadequate blood vessels growth that contributes to the worsening of a diseased state, e.g., in diseases such as coronary artery disease, stroke, and delayed wound healing. Further, ulcers, strokes, and heart attacks can result from the absence of angiogenesis that normally required for natural healing. The present invention contemplates treating those patients that are at risk of developing the above-mentioned illnesses.
[0195] Other patients that are candidates for receiving the STOP-1 antagonists of this invention have, or are at risk for developing, abnormal proliferation of fibrovascular tissue, acne rosacea, acquired immune deficiency syndrome, artery occlusion, atopic keratitis, bacterial ulcers, Bechets disease, blood borne tumors, carotid obstructive disease, choroidal neovascularization, chronic inflammation, chronic retinal detachment, chronic uveitis, chronic vitritis, contact lens overwear, corneal graft rejection, corneal neovascularization, corneal graft neovascularization, Crohn's disease, Eales disease, epidemic keratoconjunctivitis, fungal ulcers, Herpes simplex infections, Herpes zoster infections, hyperviscosity syndromes, Kaposi's sarcoma, leukemia, lipid degeneration, Lyme's disease, marginal keratolysis, Mooren ulcer, Mycobacteria infections other than leprosy, myopia, ocular neovascular disease, optic pits, Osler-Weber syndrome (Osler-Weber-Rendu, osteoarthritis, Pagets disease, pars planitis, pemphigoid, phylectenulosis, polyarteritis, post-laser complications, protozoan infections, pseudoxanthoma elasticum, pterygium keratitis sicca, radial keratotomy, retinal neovascularization, retinopathy of prematurity, retrolental fibroplasias, sarcoid, scleritis, sickle cell anemia, Sogrens syndrome, solid tumors, Stargarts disease, Steven's Johnson disease, superior limbic keratitis, syphilis, systemic lupus, Terrien's marginal degeneration, toxoplasmosis, trauma, tumors of Ewing sarcoma, tumors of neuroblastoma, tumors of osteosarcoma, tumors of retinoblastoma, tumors of rhabdomyosarcoma, ulcerative colitis, vein occlusion, Vitamin A deficiency and Wegeners sarcoidosis, undesired angiogenesis associated with diabetes, parasitic diseases, abnormal wound healing, hypertrophy following surgery, injury or trauma, inhibition of hair growth, inhibition of ovulation and corpus luteum formation, inhibition of implantation and inhibition of embryo development in the uterus.
[0196] Anti-angiogenesis therapies are useful in the general treatment of graft rejection, lung inflammation, nephrotic syndrome, preeclampsia, pericardial effusion, such as that associated with pericarditis, and pleural effusion, diseases and disorders characterized by undesirable vascular permeability, e.g., edema associated with brain tumors, ascites associated with malignancies, Meigs' syndrome, lung inflammation, nephrotic syndrome, pericardial effusion and pleural effusion and the like.
[0197] Other angiogenesis-dependent diseases that may be treated with the compositions of this invention include angiofibroma (abnormal blood of vessels which are prone to bleeding), neovascular glaucoma (abnormal growth of blood vessels in the eye), arteriovenous malformations (abnormal communication between arteries and veins), nonunion fractures (fractures that will not heal), atherosclerotic plaques (hardening of the arteries), pyogenic granuloma (common skin lesion composed of blood vessels), scleroderma (a form of connective tissue disease), hemangioma (tumor composed of blood vessels), trachoma (leading cause of blindness in the third world), hemophilic joints, vascular adhesions and hypertrophic scars (abnormal scar formation).
[0198] Since blood vessels play an important role in the regulation of bone turnover and growth, potentiators or agonists according to this invention may stimulate or enhance of bone and/or cartilage repair from disease or injury or by blocking inflammation or processes of tissue destruction (collagenase activity, osteoclast activity, etc.) mediated by inflammatory processes. Bone injuries or diseases to be treated with STOP-1 potentiators or agonists of this invention include periodontal diseases, other tooth-repair processes, osteoporosis and fractures.
[0199] A "stromal targeting agent" according to this invention is an agent that substantially recognizes and binds stromal tissue compared to other tissue. Stromal tissue is the connective tissue framework of an organ, gland, or other structure, as distinguished from the tissues performing the special function of the organ or part. Examples of stromal targeting agents include antibodies that specifically bind to FAP, fascin, HSP47, mesothelin and prostate stem antigen.
[0200] A polypeptide, antibody, antagonist or composition of this invention which "induces cell death" is one which causes a viable cell to become nonviable. The cell is one which expresses a STOP-1 polypeptide, preferably a cell that overexpresses a STOP-1 polypeptide as compared to a normal cell of the same tissue type. Preferably, the cell is a cancer cell, e.g., a breast, ovarian, stomach, endometrial, salivary gland, lung, kidney, colon, thyroid, pancreatic or bladder cell. Cell death in vitro can be determined in the absence of complement and immune effector cells to distinguish cell death induced by antibody-dependent cell-mediated cytotoxicity (ADCC) or complement dependent cytotoxicity (CDC). Thus, the assay for cell death can be performed using heat inactivated serum (i.e., in the absence of complement) and in the absence of immune effector cells. To determine whether a polypeptide, antibody, antagonist or composition of this invention is able to induce cell death, loss of membrane integrity as evaluated by uptake of propidium iodide (PI), trypan blue (see Moore et al. Cytotechnology 17:1-11 (1995)) or 7AAD can be assessed relative to untreated cells. Preferred cell death-inducing polypeptides, antibodies, antagonists or compositions are those which induce PI uptake in the PI uptake assay in BT474 cells.
[0201] A "STOP-1-expressing cell" is a cell which expresses an endogenous or transfected STOP-1 polypeptide either on the cell surface or in a secreted form. A "STOP-1-expressing cancer" is a cancer comprising cells that have a STOP-1 polypeptide present on the cell surface or that produce and secrete a STOP-1 polypeptide. In another embodiment, a "STOP-1-expressing cancer" optionally produces and secretes sufficient levels of STOP-1 polypeptide, such that a polypeptide, antibody, antagonist or composition of this invention can bind thereto and have a therapeutic effect with respect to the cancer. A cancer which "overexpresses" a STOP-1 polypeptide is one which has significantly higher levels of STOP-1 polypeptide at the cell surface thereof, or produces and secretes, compared to a noncancerous cell of the same tissue type. Such overexpression can be caused by gene amplification or by increased transcription or translation. STOP-1 polypeptide overexpression can be determined in a diagnostic or prognostic assay by evaluating increased levels of the STOP-1 protein present on the surface of a cell, or secreted by the cell (e.g., via an immunohistochemistry assay using anti-STOP-1 antibodies prepared against an isolated STOP-1 polypeptide which can be prepared using recombinant DNA technology from an isolated nucleic acid encoding the STOP-1 polypeptide; FACS analysis, etc.). Alternatively, or additionally, one can measure levels of STOP-1 polypeptide-encoding nucleic acid or mRNA in the cell, e.g., via fluorescent in situ hybridization using a nucleic acid based probe corresponding to a STOP-1-encoding nucleic acid or the complement thereof; (FISH; see WO98/45479 published October, 1998), Southern blotting, Northern blotting, or polymerase chain reaction (PCR) techniques, such as real time quantitative PCR (RT-PCR). One can also study STOP-1 polypeptide overexpression by measuring shed antigen in a biological fluid such as serum, e.g., using antibody-based assays (see also, e.g., U.S. Pat. No. 4,933,294 issued Jun. 12, 1990; WO91/05264 published Apr. 18, 1991; U.S. Pat. No. 5,401,638 issued Mar. 28, 1995; and Sias et al., J. Immunol. Methods 132:73-80 (1990)). Aside from the above assays, various in vivo assays are available to the skilled practitioner. For example, one can expose cells within the body of the mammal to an antibody which is optionally labeled with a detectable label, e.g., a radioactive isotope, and binding of the antibody to cells in the mammal can be evaluated, e.g., by external scanning for radioactivity or by analyzing a biopsy taken from a mammal previously exposed to the antibody.
[0202] The word "label" when used herein refers to a detectable compound or composition which is conjugated directly or indirectly to the polypeptide, antibody, antagonist or composition so as to generate a "labeled" a polypeptide, antibody, antagonist or composition. The label can be detectable by itself (e.g. radioisotope labels or fluorescent labels) or, in the case of an enzymatic label, can catalyze chemical alteration of a substrate compound or composition which is detectable.
[0203] The term "cytotoxic agent" as used herein refers to a substance that inhibits or prevents the function of cells and/or causes destruction of cells. The term is intended to include radioactive isotopes (e.g., At211, I131, I125, Y90, Re186, Re188, Sm153, Bi212, P32 and radioactive isotopes of Lu), chemotherapeutic agents e.g. methotrexate, adriamicin, vinca alkaloids (vincristine, vinblastine, etoposide), doxorubicin, melphalan, mitomycin C, chlorambucil, daunorubicin or other intercalating agents, enzymes and fragments thereof such as nucleolytic enzymes, antibiotics, and toxins such as small molecule toxins or enzymatically active toxins of bacterial, fungal, plant or animal origin, including fragments and/or variants thereof, and the various antitumor or anticancer agents disclosed below. Other cytotoxic agents are described below. A tumoricidal agent causes destruction of tumor cells.
[0204] A "chemotherapeutic agent" is a chemical compound useful in the treatment of cancer. Examples of chemotherapeutic agents include alkylating agents such as thiotepa and CYTOXAN® cyclosphosphamide; alkyl sulfonates such as busulfan, improsulfan and piposulfan; aziridines such as benzodopa, carboquone, meturedopa, and uredopa; ethylenimines and methylamelamines including altretamine, triethylenemelamine, trietylenephosphoramide, triethiylenethiophosphoramide and trimethylolomelamine; acetogenins (especially bullatacin and bullatacinone); a camptothecin (including the synthetic analogue topotecan); bryostatin; callystatin; CC-1065 (including its adozelesin, carzelesin and bizelesin synthetic analogues); cryptophycins (particularly cryptophycin 1 and cryptophycin 8); dolastatin; duocarmycin (including the synthetic analogues, KW-2189 and CB1-TM1); eleutherobin; pancratistatin; a sarcodictyin; spongistatin; nitrogen mustards such as chlorambucil, chlornaphazine, cholophosphamide, estramustine, ifosfamide, mechlorethamine, mechlorethamine oxide hydrochloride, melphalan, novembichin, phenesterine, prednimustine, trofosfamide, uracil mustard; nitrosureas such as carmustine, chlorozotocin, fotemustine, lomustine, nimustine, and ranimnustine; antibiotics such as the enediyne antibiotics (e.g., calicheamicin, especially calicheamicin gamma1I and calicheamicin omegaI1 (see, e.g., Agnew, Chem. Intl. Ed. Engl., 33: 183-186 (1994)); dynemicin, including dynemicin A; bisphosphonates, such as clodronate; an esperamicin; as well as neocarzinostatin chromophore and related chromoprotein enediyne antiobiotic chromophores), aclacinomysins, actinomycin, authramycin, azaserine, bleomycins, cactinomycin, carabicin, caminomycin, carzinophilin, chromomycinis, dactinomycin, daunorubicin, detorubicin, 6-diazo-5-oxo-L-norleucine, ADRIAMYCIN® doxorubicin (including morpholino-doxorubicin, cyanomorpholino-doxorubicin, 2-pyrrolino-doxorubicin and deoxydoxorubicin), epirubicin, esorubicin, idarubicin, marcellomycin, mitomycins such as mitomycin C, mycophenolic acid, nogalamycin, olivomycins, peplomycin, potfiromycin, puromycin, quelamycin, rodorubicin, streptonigrin, streptozocin, tubercidin, ubenimex, zinostatin, zorubicin; anti-metabolites such as methotrexate and 5-fluorouracil (5-FU); folic acid analogues such as denopterin, methotrexate, pteropterin, trimetrexate; purine analogs such as fludarabine, 6-mercaptopurine, thiamiprine, thioguanine; pyrimidine analogs such as ancitabine, azacitidine, 6-azauridine, carmofur, cytarabine, dideoxyuridine, doxifluridine, enocitabine, floxuridine; androgens such as calusterone, dromostanolone propionate, epitiostanol, mepitiostane, testolactone; anti-adrenals such as aminoglutethimide, mitotane, trilostane; folic acid replenisher such as frolinic acid; aceglatone; aldophosphamide glycoside; aminolevulinic acid; eniluracil; amsacrine; bestrabucil; bisantrene; edatraxate; defofamine; demecolcine; diaziquone; elformithine; elliptinium acetate; an epothilone; etoglucid; gallium nitrate; hydroxyurea; lentinan; lonidainine; maytansinoids such as maytansine and ansamitocins; mitoguazone; mitoxantrone; mopidanmol; nitraerine; pentostatin; phenamet; pirarubicin; losoxantrone; podophyllinic acid; 2-ethylhydrazide; procarbazine; PSK® polysaccharide complex (JHS Natural Products, Eugene, Oreg.); razoxane; rhizoxin; sizofuran; spirogermanium; tenuazonic acid; triaziquone; 2,2',2''-trichlorotriethylamine; trichothecenes (especially T-2 toxin, verracurin A, roridin A and anguidine); urethan; vindesine; dacarbazine; mannomustine; mitobronitol; mitolactol; pipobroman; gacytosine; arabinoside ("Ara-C"); cyclophosphamide; thiotepa; taxoids, e.g., TAXOL® paclitaxel (Bristol-Myers Squibb Oncology, Princeton, N.J.), ABRAXANE® Cremophor-free, albumin-engineered nanoparticle formulation of paclitaxel (American Pharmaceutical Partners, Schaumberg, Ill.), and TAXOTERE® doxetaxel (Rhone-Poulenc Rorer, Antony, France); chloranbucil; GEMZAR® gemcitabine; 6-thioguanine; mercaptopurine; methotrexate; platinum analogs such as cisplatin and carboplatin; vinblastine; platinum; etoposide (VP-16); ifosfamide; mitoxantrone; vincristine; NAVELBINE® vinorelbine; novantrone; teniposide; edatrexate; daunomycin; aminopterin; xeloda; ibandronate; CPT-11; topoisomerase inhibitor RFS 2000; difluoromethylornithine (DMFO); retinoids such as retinoic acid; capecitabine; and pharmaceutically acceptable salts, acids or derivatives of any of the above.
[0205] Also included in this definition are anti-hormonal agents that act to regulate or inhibit hormone action on tumors such as anti-estrogens and selective estrogen receptor modulators (SERMs), including, for example, tamoxifen (including NOLVADEX® tamoxifen), raloxifene, droloxifene, 4-hydroxytamoxifen, trioxifene, keoxifene, LY117018, onapristone, and FARESTON•toremifene; aromatase inhibitors that inhibit the enzyme aromatase, which regulates estrogen production in the adrenal glands, such as, for example, 4(5)-imidazoles, aminoglutethimide, MEGASE® megestrol acetate, AROMASIN® exemestane, formestanie, fadrozole, RIVISOR® vorozole, FEMARA® letrozole, and ARIMIDEX® anastrozole; and anti-androgens such as flutamide, nilutamide, bicalutamide, leuprolide, and goserelin; as well as troxacitabine (a 1,3-dioxolane nucleoside cytosine analog); antisense oligonucleotides, particularly those which inhibit expression of genes in signaling pathways implicated in abherant cell proliferation, such as, for example, PKC-alpha, Ralf and H-Ras; ribozymes such as a VEGF expression inhibitor (e.g., ANGIOZYME® ribozyme) and a HER2 expression inhibitor; vaccines such as gene therapy vaccines, for example, ALLOVECTIN® vaccine, LEUVECTIN® vaccine, and VAXID® vaccine; PROLEUKIN® rIL-2; LURTOTECAN® topoisomerase 1 inhibitor; ABARELIX® rmRH; and pharmaceutically acceptable salts, acids or derivatives of any of the above.
[0206] A "growth inhibitory agent" when used herein refers to a compound or composition which inhibits growth of a cell, especially a STOP-1-expressing cancer cell, either in vitro or in vivo. Thus, the growth inhibitory agent can be one which significantly reduces the percentage of STOP-1-expressing cells in S phase. Examples of growth inhibitory agents include agents that block cell cycle progression (at a place other than S phase), such as agents that induce G1 arrest and M-phase arrest. Classical M-phase blockers include the vincas (vincristine and vinblastine), taxanes, and topoisomerase II inhibitors such as doxorubicin, epirubicin, daunorubicin, etoposide, and bleomycin. Those agents that arrest G1 also spill over into S-phase arrest, for example, DNA alkylating agents such as tamoxifen, prednisone, dacarbazine, mechlorethamine, cisplatin, methotrexate, 5-fluorouracil, and ara-C. Further information can be found in The Molecular Basis of Cancer, Mendelsohn and Israel, eds., Chapter 1, entitled "Cell cycle regulation, oncogenes, and antineoplastic drugs" by Murakami et al. (WB Saunders: Philadelphia, 1995), especially p. 13. The taxanes (paclitaxel and docetaxel) are anticancer drugs both derived from the yew tree. Docetaxel (TAXOTERE®, Rhone-Poulenc Rorer), derived from the European yew, is a semisynthetic analogue of paclitaxel (TAXOL®, Bristol-Myers Squibb). Paclitaxel and docetaxel promote the assembly of microtubules from tubulin dimers and stabilize microtubules by preventing depolymerization, which results in the inhibition of mitosis in cells.
[0207] "Doxorubicin" is an anthracycline antibiotic. The full chemical name of doxorubicin is (8S-cis)-10-[(3-amino-2,3,6-trideoxy-L-lyxo-hexapyranosyl)oxy]-7,8,9,10-t- etrahydro-6, 8,11-trihydroxy-8-(hydroxyacetyl)-1-methoxy-5,12-naphthacenedione.
[0208] The term "package insert" is used to refer to instructions customarily included in commercial packages of therapeutic products, that contain information about the indications, usage, dosage, administration, contraindications and/or warnings concerning the use of such therapeutic products.
TABLE-US-00007 TABLE 1 /* * * C-C increased from 12 to 15 * Z is average of EQ * B is average of ND * match with stop is _M; stop-stop = 0; J (joker) match = 0 */ #define _M -8 /* value of a match with a stop */ int _day[26][26] = { /* A B C D E F G H I J K L M N O P Q R S T U V W X Y Z */ /* A */ { 2, 0,-2, 0, 0,-4, 1,-1,-1, 0,-1,-2,-1, 0,_M, 1, 0,-2, 1, 1, 0, 0,-6, 0,-3, 0}, /* B */ { 0, 3,-4, 3, 2,-5, 0, 1,-2, 0, 0,-3,-2, 2,_M,-1, 1, 0, 0, 0, 0,-2,-5, 0,-3, 1}, /* C */ {-2,-4,15,-5,-5,-4,-3,-3,-2, 0,-5,-6,-5,-4,_M,-3,-5,-4, 0,-2, 0,-2,-8, 0, 0,-5}, /* D */ { 0, 3,-5, 4, 3,-6, 1, 1,-2, 0, 0,-4,-3, 2,_M,-1, 2,-1, 0, 0, 0,-2,-7, 0,-4, 2}, /* E */ { 0, 2,-5, 3, 4,-5, 0, 1,-2, 0, 0,-3,-2, 1,_M,-1, 2,-1, 0, 0, 0,-2,-7, 0,-4, 3}, /* F */ {-4,-5,-4,-6,-5, 9,-5,-2, 1, 0,-5, 2, 0,-4,_M,-5,-5,-4,-3,-3, 0,-1, 0, 0, 7,-5}, /* G */ { 1, 0,-3, 1, 0,-5, 5,-2,-3, 0,-2,-4,-3, 0,_M,-1,-1,-3, 1, 0, 0,-1,-7, 0,-5, 0}, /* H */ {-1, 1,-3, 1, 1,-2,-2, 6,-2, 0, 0,-2,-2, 2,_M, 0, 3, 2,-1,-1, 0,-2,-3, 0, 0, 2}, /* I */ {-1,-2,-2,-2,-2, 1,-3,-2, 5, 0,-2, 2, 2,-2,_M,-2,-2,-2,-1, 0, 0, 4,-5, 0,-1,-2}, /* J */ { 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,_M, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0}, /* K */ {-1, 0,-5, 0, 0,-5,-2, 0,-2, 0, 5,-3, 0, 1,_M,-1, 1, 3, 0, 0, 0,-2,-3, 0,-4, 0}, /* L */ {-2,-3,-6,-4,-3, 2,-4,-2, 2, 0,-3, 6, 4,-3,_M,-3,-2,-3,-3,-1, 0, 2,-2, 0,-1,-2}, /* M */ {-1,-2,-5,-3,-2, 0,-3,-2, 2, 0, 0, 4, 6,-2,_M,-2,-1, 0,-2,-1, 0, 2,-4, 0,-2,-1}, /* N */ { 0, 2,-4, 2, 1,-4, 0, 2,-2, 0, 1,-3,-2, 2,_M,-1, 1, 0, 1, 0, 0,-2,-4, 0,-2, 1}, /* O */ {_M,_M,_M,_M,_M,_M,_M,_M,_M,_M,_M,_M,_M,_M, 0,_M,_M,_M,_M,_M,_M,_M,_M,_M,_M,_M}, /* P */ { 1,-1,-3,-1,-1,-5,-1, 0,-2, 0,-1,-3,-2,-1,_M, 6, 0, 0, 1, 0, 0,-1,-6, 0,-5, 0}, /* Q */ { 0, 1,-5, 2, 2,-5,-1, 3,-2, 0, 1,-2,-1, 1,_M, 0, 4, 1,-1,-1, 0,-2,-5, 0,-4, 3}, /* R */ {-2, 0,-4,-1,-1,-4,-3, 2,-2, 0, 3,-3, 0, 0,_M, 0, 1, 6, 0,-1, 0,-2, 2, 0,-4, 0}, /* S */ { 1, 0, 0, 0, 0,-3, 1,-1,-1, 0, 0,-3,-2, 1,_M, 1,-1, 0, 2, 1, 0,-1,-2, 0,-3, 0}, /* T */ { 1, 0,-2, 0, 0,-3, 0,-1, 0, 0, 0,-1,-1, 0,_M, 0,-1,-1, 1, 3, 0, 0,-5, 0,-3, 0}, /* U */ { 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,_M, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0}, /* V */ { 0,-2,-2,-2,-2,-1,-1,-2, 4, 0,-2, 2, 2,-2,_M,-1,-2,-2,-1, 0, 0, 4,-6, 0,-2,-2}, /* W */ {-6,-5,-8,-7,-7, 0,-7,-3,-5, 0,-3,-2,-4,-4,_M,-6,-5, 2,-2,-5, 0,-6,17, 0, 0,-6}, /* X */ { 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,_M, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0}, /* Y */ {-3,-3, 0,-4,-4, 7,-5, 0,-1, 0,-4,-1,-2,-2,_M,-5,-4,-4,-3,-3, 0,-2, 0, 0,10,-4}, /* Z */ { 0, 1,-5, 2, 3,-5, 0, 2,-2, 0, 0,-2,-1, 1,_M, 0, 3, 0, 0, 0, 0,-2,-6, 0,-4, 4} }; /* */ #include <stdio.h> #include <ctype.h> #define MAXJMP 16 /* max jumps in a diag */ #define MAXGAP 24 /* don't continue to penalize gaps larger than this */ #define JMPS 1024 /* max jmps in an path */ #define MX 4 /* save if there's at least MX-1 bases since last jmp */ #define DMAT 3 /* value of matching bases */ #define DMIS 0 /* penalty for mismatched bases */ #define DINS0 8 /* penalty for a gap */ #define DINS1 1 /* penalty per base */ #define PINS0 8 /* penalty for a gap */ #define PINS1 4 /* penalty per residue */ struct jmp { short n[MAXJMP]; /* size of jmp (neg for dely) */ unsigned short x[MAXJMP]; /* base no. of jmp in seq x */ }; /* limits seq to 2{circumflex over ( )}16 -1 */ struct diag { int score; /* score at last jmp */ long offset; /* offset of prev block */ short ijmp; /* current jmp index */ struct jmp jp; /* list of jmps */ }; struct path { int spc; /* number of leading spaces */ short n[JMPS]; /* size of jmp (gap) */ int x[JMPS]; /* loc of jmp (last elem before gap) */ }; char *ofile; /* output file name */ char *namex[2]; /* seq names: getseqs( ) */ char *prog; /* prog name for err msgs */ char *seqx[2]; /* seqs: getseqs( ) */ int dmax; /* best diag: nw( ) */ int dmax( ); /* final diag */ int dna; /* set if dna: main( ) */ int endgaps; /* set if penalizing end gaps */ int gapx, gapy; /* total gaps in seqs */ int len0, len1; /* seq lens */ int ngapx, ngapy; /* total size of gaps */ int smax; /* max score: nw( ) */ int *xbm; /* bitmap for matching */ long offset; /* current offset in jmp file */ struct diag *dx; /* holds diagonals */ struct path pp[2]; /* holds path for seqs */ char *calloc( ), *malloc( ), *index( ), *strcpy( ); char *getseq( ), *g_calloc( ); /* Needleman-Wunsch alignment program * * usage: progs file1 file2 * where file1 and file2 are two dna or two protein sequences. * The sequences can be in upper- or lower-case an may contain ambiguity * Any lines beginning with `;`, `>` or `<` are ignored * Max file length is 65535 (limited by unsigned short x in the jmp struct) * A sequence with 1/3 or more of its elements ACGTU is assumed to be DNA * Output is in the file "align.out" * * The program may create a tmp file in /tmp to hold info about traceback. * Original version developed under BSD 4.3 on a vax 8650 */ #include "nw.h" #include "day.h" static _dbval[26] = { 1,14,2,13,0,0,4,11,0,0,12,0,3,15,0,0,0,5,6,8,8,7,9,0,10,0 }; static _pbval[26] = { 1, 2|(1<<(`D`-`A`))|(1<<(`N`-`A`)), 4, 8, 16, 32, 64, 128, 256, 0xFFFFFFF, 1<<10, 1<<11, 1<<12, 1<<13, 1<<14, 1<<15, 1<<16, 1<<17, 1<<18, 1<<19, 1<<20, 1<<21, 1<<22, 1<<23, 1<<24, 1<<25|(1<<(`E`-`A`))|(1<<(`Q`-`A`)) }; main(ac, av) main int ac; char *av[ ]; { prog = av[0]; if (ac != 3) { fprintf(stderr,"usage: %s file1 file2\n", prog); fprintf(stderr,"where file1 and file2 are two dna or two protein sequences.\n"); fprintf(stderr,"The sequences can be in upper- or lower-case\n"); fprintf(stderr,"Any lines beginning with `;` or `<` are ignored\n"); fprintf(stderr,"Output is in the file \"align.out\"\n"); exit(1); } namex[0] = av[1]; namex[1] = av[2]; seqx[0] = getseq(namex[0], &len0); seqx[1] = getseq(namex[1], &len1); xbm = (dna)? _dbval : _pbval; endgaps = 0; /* 1 to penalize endgaps */ ofile = "align.out"; /* output file */ nw( ); /* fill in the matrix, get the possible jmps */ readjmps( ); /* get the actual jmps */ print( ); /* print stats, alignment */ cleanup(0); /* unlink any tmp files */ } /* do the alignment, return best score: main( ) * dna: values in Fitch and Smith, PNAS, 80, 1382-1386, 1983 * pro: PAM 250 values * When scores are equal, we prefer mismatches to any gap, prefer * a new gap to extending an ongoing gap, and prefer a gap in seqx * to a gap in seq y. */ nw( ) nw { char *px, *py; /* seqs and ptrs */ int *ndely, *dely; /* keep track of dely */ int ndelx, delx; /* keep track of delx */ int *tmp; /* for swapping row( ), row1 */ int mis; /* score for each type */ int ins0, ins1; /* insertion penalties */ register id; /* diagonal index */ register ij; /* jmp index */ register *col0, *col1; /* score for curr, last row */ register xx, yy; /* index into seqs */ dx = (struct diag *)g_calloc("to get diags", len0+len1+1, sizeof(struct diag)); ndely = (int *)g_calloc("to get ndely", len1+1, sizeof(int)); dely = (int *)g_calloc("to get dely", len1+1, sizeof(int)); col0 = (int *)g_calloc("to get col0", len1+1, sizeof(int)); col1 = (int *)g_calloc("to get col1", len1+1, sizeof(int)); ins0 = (dna)? DINS0 : PINS0; ins1 = (dna)? DINS1 : PINS1; smax = -10000; if (endgaps) { for (col0[0] = dely[0] = -ins0, yy = 1; yy <= len1; yy++) { col0[yy] = dely[yy] = col0[yy-1] - ins1; ndely[yy] = yy; } col0[0] = 0; /* Waterman Bull Math Biol 84 */ } else for (yy = 1; yy <= len1; yy++) dely[yy] = -ins0; /* fill in match matrix */ for (px = seqx[0], xx = 1; xx <= len0; px++, xx++) { /* initialize first entry in col */ if (endgaps) { if (xx == 1) col1[0] = delx = -(ins0+ins1); else col1[0] = delx = col0[0] - ins1; ndelx = xx; } else { col1[0] = 0; delx = -ins0; ndelx = 0; } ...nw for (py = seqx[1], yy = 1; yy <= len1; py++, yy++) { mis = col0[yy-1]; if (dna) mis += (xbm[*px-`A`]&xbm[*py-`A`])? DMAT : DMIS; else mis += _day[*px-`A`][*py-`A`]; /* update penalty for del in x seq; * favor new del over ongong del * ignore MAXGAP if weighting endgaps */ if (endgaps || ndely[yy] < MAXGAP) { if (col0[yy] - ins0 >= dely[yy]) { dely[yy] = col0[yy] - (ins0+ins1); ndely[yy] = 1; } else { dely[yy] -= ins1; ndely[yy]++; } } else { if (col0[yy] - (ins0+ins1) >= dely[yy]) { dely[yy] = col0[yy] - (ins0+ins1); ndely[yy] = 1; } else ndely[yy]++; } /* update penalty for del in y seq; * favor new del over ongong del
*/ if (endgaps || ndelx < MAXGAP) { if (col1[yy-1] - ins0 >= delx) { delx = col1[yy-1] - (ins0+ins1); ndelx = 1; } else { delx -= ins1; ndelx++; } } else { if (col1[yy-1] - (ins0+ins1) >= delx) { delx = col1[yy-1] - (ins0+ins1); ndelx = 1; } else ndelx++; } /* pick the maximum score; we're favoring * mis over any del and delx over dely */ ...nw id = xx - yy + len1 - 1; if (mis >= delx && mis >= dely[yy]) col1[yy] = mis; else if (delx >= dely[yy]) { col1[yy] = delx; ij = dx[id].ijmp; if (dx[id].jp.n[0] && (!dna || (ndelx >= MAXJMP && xx > dx[id].jp.x[ij]+MX) || mis > dx[id].score+DINS0)) { dx[id].ijmp++; if (++ij >= MAXJMP) { writejmps(id); ij = dx[id].ijmp = 0; dx[id].offset = offset; offset += sizeof(struct jmp) + sizeof(offset); } } dx[id].jp.n[ij] = ndelx; dx[id].jp.x[ij] = xx; dx[id].score = delx; } else { col1[yy] = dely[yy]; ij = dx[id].ijmp; if (dx[id].jp.n[0] && (!dna || (ndely[yy] >= MAXJMP && xx > dx[id].jp.x[ij]+MX) || mis > dx[id].score+DINS0)) { dx[id].ijmp++; if (++ij >= MAXJMP) { writejmps(id); ij = dx[id].ijmp = 0; dx[id].offset = offset; offset += sizeof(struct jmp) + sizeof(offset); } } dx[id].jp.n[ij] = -ndely[yy]; dx[id].jp.x[ij] = xx; dx[id].score = dely[yy]; } if (xx == len0 && yy < len1) { /* last col */ if (endgaps) col1[yy] -= ins0+ins1*(len1-yy); if (col1[yy] > smax) { smax = col1[yy]; dmax = id; } } } if (endgaps && xx < len0) col1[yy-1] -= ins0+ins1*(len0-xx); if (col1[yy-1] > smax) { smax = col1[yy-1]; dmax = id; } tmp = col0; col0 = col1; col1 = tmp; } (void) free((char *)ndely); (void) free((char *)dely); (void) free((char *)col0); (void) free((char *)col1); } /* * * print( ) -- only routine visible outside this module * * static: * getmat( ) -- trace back best path, count matches: print( ) * pr_align( ) -- print alignment of described in array p[ ]: print( ) * dumpblock( ) -- dump a block of lines with numbers, stars: pr_align( ) * nums( ) -- put out a number line: dumpblock( ) * putline( ) -- put out a line (name, [num], seq, [num]): dumpblock( ) * stars( ) - -put a line of stars: dumpblock( ) * stripname( ) -- strip any path and prefix from a seqname */ #include "nw.h" #define SPC 3 #define P_LINE 256 /* maximum output line */ #define P_SPC 3 /* space between name or num and seq */ extern _day[26][26]; int olen; /* set output line length */ FILE *fx; /* output file */ print( ) print { int lx, ly, firstgap, lastgap; /* overlap */ if ((fx = fopen(ofile, "w")) == 0) { fprintf(stderr,"%s: can't write %s\n", prog, ofile); cleanup(1); } fprintf(fx, "<first sequence: %s (length = %d)\n", namex[0], len0); fprintf(fx, "<second sequence: %s (length = %d)\n", namex[1], len1); olen = 60; lx = len0; ly = len1; firstgap = lastgap = 0; if (dmax < len1 - 1) { /* leading gap in x */ pp[0].spc = firstgap = len1 - dmax - 1; ly -= pp[0].spc; } else if (dmax > len1 - 1) { /* leading gap in y */ pp[1].spc = firstgap = dmax - (len1 - 1); lx -= pp[1].spc; } if (dmax0 < len0 - 1) { /* trailing gap in x */ lastgap = len0 - dmax0 -1; lx -= lastgap; } else if (dmax0 > len0 - 1) { /* trailing gap in y */ lastgap = dmax0 - (len0 - 1); ly -= lastgap; } getmat(lx, ly, firstgap, lastgap); pr_align( ); } /* * trace back the best path, count matches */ static getmat(lx, ly, firstgap, lastgap) getmat int lx, ly; /* "core" (minus endgaps) */ int firstgap, lastgap; /* leading trailing overlap */ { int nm, i0, i1, siz0, siz1; char outx[32]; double pct; register n0, n1; register char *p0, *p1; /* get total matches, score */ i0 = i1 = siz0 = siz1 = 0; p0 = seqx[0] + pp[1].spc; p1 = seqx[1] + pp[0].spc; n0 = pp[1].spc + 1; n1 = pp[0].spc + 1; nm = 0; while ( *p0 && *p1 ) { if (siz0) { p1++; n1++; siz0--; } else if (siz1) { p0++; n0++; siz1--; } else { if (xbm[*p0-`A`]&xbm[*p1-`A`]) nm++; if (n0++ == pp[0].x[i0]) siz0 = pp[0].n[i0++]; if (n1++ == pp[1].x[i1]) siz1 = pp[1].n[i1++]; p0++; p1++; } } /* pct homology: * if penalizing endgaps, base is the shorter seq * else, knock off overhangs and take shorter core */ if (endgaps) lx = (len0 < len1)? len0 : len1; else lx = (lx < ly)? lx : ly; pct = 100.*(double)nm/(double)lx; fprintf(fx, "\n"); fprintf(fx, "<%d match%s in an overlap of %d: %.2f percent similarity\n", nm, (nm == 1)? "" : "es", lx, pct); fprintf(fx, "<gaps in first sequence: %d", gapx); ...getmat if (gapx) { (void) sprintf(outx, " (%d %s%s)", ngapx, (dna)? "base":"residue", (ngapx == 1)? "":"s"); fprintf(fx,"%s", outx); fprintf(fx, ", gaps in second sequence: %d", gapy); if (gapy) { (void) sprintf(outx, " (%d %s%s)", ngapy, (dna)? "base":"residue", (ngapy == 1)? "":"s"); fprintf(fx,"%s", outx); } if (dna) fprintf(fx, "\n<score: %d (match = %d, mismatch = %d, gap penalty = %d + %d per base)\n", smax, DMAT, DMIS, DINS0, DINS1); else fprintf(fx, "\n<score: %d (Dayhoff PAM 250 matrix, gap penalty = %d + %d per residue)\n", smax, PINS0, PINS1); if (endgaps) fprintf(fx, "<endgaps penalized. left endgap: %d %s%s, right endgap: %d %s%s\n", firstgap, (dna)? "base" : "residue", (firstgap == 1)? "" : "s", lastgap, (dna)? "base" : "residue", (lastgap == 1)? "" : "s"); else fprintf(fx, "<endgaps not penalized\n"); } static nm; /* matches in core -- for checking */ static lmax; /* lengths of stripped file names */ static ij[2]; /* jmp index for a path */ static nc[2]; /* number at start of current line */ static ni[2]; /* current elem number -- for gapping */ static siz[2]; static char *ps[2]; /* ptr to current element */ static char *po[2]; /* ptr to next output char slot */ static char out[2][P_LINE]; /* output line */ static char star[P_LINE]; /* set by stars( ) */ /* * print alignment of described in struct path pp[ ] */ static pr_align( ) pr_align { int nn; /* char count */ int more; register i; for (i = 0, lmax = 0; i < 2; i++) { nn = stripname(namex[i]); if (nn > lmax) lmax = nn; nc[i] = 1; ni[i] = 1; siz[i] = ij[i] = 0; ps[i] = seqx[i]; po[i] = out[i]; } for (nn = nm = 0, more = 1; more; ) { ...pr_align for (i = more = 0; i < 2; i++) { /* * do we have more of this sequence? */ if (!*ps[i])
continue; more++; if (pp[i].spc) { /* leading space */ *po[i]++ = ` `; pp[i].spc--; } else if (siz[i]) { /* in a gap */ *po[i]++ = `-`; siz[i]--; } else { /* we're putting a seq element */ *po[i] = *ps[i]; if (islower(*ps[i])) *ps[i] = toupper(*ps[i]); po[i]++; ps[i]++; /* * are we at next gap for this seq? */ if (ni[i] == pp[i].x[ij[i]]) { /* * we need to merge all gaps * at this location */ siz[i] = pp[i].n[ij[i]++]; while (ni[i] == pp[i].x[ij[i]]) siz[i] += pp[i].n[ij[i]++]; } ni[i]++; } } if (++nn == olen || !more && nn) { dumpblock( ); for (i = 0; i < 2; i++) po[i] = out[i]; nn = 0; } } } /* * dump a block of lines, including numbers, stars: pr_align( ) */ static dumpblock( ) dumpblock { register i; for (i = 0; i < 2; i++) *po[i]-- = `\0`; ...dumpblock (void) putc(`\n`, fx); for (i = 0; i < 2; i++) { if (*out[i] && (*out[i] != ` ` || *(po[i]) != ` `)) { if (i == 0) nums(i); if (i == 0 && *out[1]) stars( ); putline(i); if (i == 0 && *out[1]) fprintf(fx, star); if (i == 1) nums(i); } } } /* * put out a number line: dumpblock( ) */ static nums(ix) nums int ix; /* index in out[ ] holding seq line */ { char nline[P_LINE]; register i, j; register char *pn, *px, *py; for (pn = nline, i = 0; i < lmax+P_SPC; i++, pn++) *pn = ` `; for (i = nc[ix], py = out[ix]; *py; py++, pn++) { if (*py == ` ` || *py == `-`) *pn = ` `; else { if (i%10 == 0 || (i == 1 && nc[ix] != 1)) { j = (i < 0)? -i : i; for (px = pn; j; j /= 10, px--) *px = j%10 + `0`; if (i < 0) *px = `-`; } else *pn = ` `; i++; } } *pn = `\0`; nc[ix] = i; for (pn = nline; *pn; pn++) (void) putc(*pn, fx); (void) putc(`\n`, fx); } /* * put out a line (name, [num], seq, [num]): dumpblock( ) */ static putline(ix) putline int ix; { ...putline int i; register char *px; for (px = namex[ix], i = 0; *px && *px != `:`; px++, i++) (void) putc(*px, fx); for (; i < lmax+P_SPC; i++) (void) putc(` `, fx); /* these count from 1: * ni[ ] is current element (from 1) * nc[ ] is number at start of current line */ for (px = out[ix]; *px; px++) (void) putc(*px&0x7F, fx); (void) putc(`\n`, fx); } /* * put a line of stars (seqs always in out[0], out[1]): dumpblock( ) */ static stars( ) stars { int i; register char *p0, *p1, cx, *px; if (!*out[0] || (*out[0] == ` ` && *(po[0]) == ` `) || .sup. !*out[1] || (*out[1] == ` ` && *(po[1]) == ` `)) return; px = star; for (i = lmax+P_SPC; i; i--) *px++ = ` `; for (p0 = out[0], p1 = out[1]; *p0 && *p1; p0++, p1++) { if (isalpha(*p0) && isalpha(*p1)) { if (xbm[*p0-`A`]&xbm[*p1-`A`]) { cx = `*`; nm++; } else if (!dna && _day[*p0-`A`][*p1-`A`] > 0) cx = `.`; else cx = ` `; } else cx = ` `; *px++ = cx; } *px++ = `\n`; *px = `\0`; } /* * strip path or prefix from pn, return len: pr_align( ) */ static stripname(pn) stripname char *pn; /* file name (may be path) */ { register char *px, *py; py = 0; for (px = pn; *px; px++) if (*px == `/`) py = px + 1; if (py) (void) strcpy(pn, py); return(strlen(pn)); } /* * cleanup( ) -- cleanup any tmp file * getseq( ) -- read in seq, set dna, len, maxlen * g_calloc( ) -- calloc( ) with error checkin * readjmps( ) -- get the good jmps, from tmp file if necessary * writejmps( ) -- write a filled array of jmps to a tmp file: nw( ) */ #include "nw.h" #include <sys/file.h> char *jname = "/tmp/homgXXXXXX"; /* tmp file for jmps */ FILE *fj; int cleanup( ); /* cleanup tmp file */ long lseek( ); /* * remove any tmp file if we blow */ cleanup(i) cleanup int i; { if (fj) (void) unlink(jname); exit(i); } /* * read, return ptr to seq, set dna, len, maxlen * skip lines starting with `;`, `<`, or `>` * seq in upper or lower case */ char * getseq(file, len) getseq char *file; /* file name */ int *len; /* seq len */ { char line[1024], *pseq; register char *px, *py; int natgc, tlen; FILE *fp; if ((fp = fopen(file,"r")) == 0) { fprintf(stderr,"%s: can't read %s\n", prog, file); exit(1); } tlen = natgc = 0; while (fgets(line, 1024, fp)) { if (*line == `;` || *line == `<` || *line == `>`) continue; for (px = line; *px != `\n`; px++) if (isupper(*px) || islower(*px)) tlen++; } if ((pseq = malloc((unsigned)(tlen+6))) == 0) { fprintf(stderr,"%s: malloc( ) failed to get %d bytes for %s\n", prog, tlen+6, file); exit(1); } pseq[0] = pseq[1] = pseq[2] = pseq[3] = `\0`; ...getseq py = pseq + 4; *len = tlen; rewind(fp); while (fgets(line, 1024, fp)) { if (*line == `;` || *line == `<` || *line == `>`) continue; for (px = line; *px != `\n`; px++) { if (isupper(*px)) *py++ = *px; else if (islower(*px)) *py++ = toupper(*px); if (index("ATGCU",*(py-1))) natgc++; } } *py++ = `\0`; *py = `\0`; (void) fclose(fp); dna = natgc > (tlen/3); return(pseq+4); } char * g_calloc(msg, nx, sz) g_calloc char *msg; /* program, calling routine */ int nx, sz; /* number and size of elements */ {
char *px, *calloc( ); if ((px = calloc((unsigned)nx, (unsigned)sz)) == 0) { if (*msg) { fprintf(stderr, "%s: g_calloc( ) failed %s (n=%d, sz=%d)\n", prog, msg, nx, sz); exit(1); } } return(px); } /* * get final jmps from dx[ ] or tmp file, set pp[ ], reset dmax: main( ) */ readjmps( ) readjmps { int fd = -1; int siz, i0, i1; register i, j, xx; if (fj) { (void) fclose(fj); if ((fd = open(jname, O_RDONLY, 0)) < 0) { fprintf(stderr, "%s: can't open( ) %s\n", prog, jname); cleanup(1); } } for (i = i0 = i1 = 0, dmax0 = dmax, xx = len0; ; i++) { while (1) { for (j = dx[dmax].ijmp; j >= 0 && dx[dmax].jp.x[j] >= xx; j--) ; ...readjmps if (j < 0 && dx[dmax].offset && fj) { (void) lseek(fd, dx[dmax].offset, 0); (void) read(fd, (char *)&dx[dmax].jp, sizeof(struct jmp)); (void) read(fd, (char *)&dx[dmax].offset, sizeof(dx[dmax].offset)); dx[dmax].ijmp = MAXJMP-1; } else break; } if (i >= JMPS) { fprintf(stderr, "%s: too many gaps in alignment\n", prog); cleanup(1); } if (j >= 0) { siz = dx[dmax].jp.n[j]; xx = dx[dmax].jp.x[j]; dmax += siz; if (siz < 0) { /* gap in second seq */ pp[1].n[i1] = -siz; xx += siz; /* id = xx - yy + len1 - 1 */ pp[1].x[i1] = xx - dmax + len1 - 1; gapy++; ngapy -= siz; /* ignore MAXGAP when doing endgaps */ siz = (-siz < MAXGAP || endgaps)? -siz : MAXGAP; i1++; } else if (siz > 0) { /* gap in first seq */ pp[0].n[i0] = siz; pp[0].x[i0] = xx; gapx++; ngapx += siz; /* ignore MAXGAP when doing endgaps */ siz = (siz < MAXGAP || endgaps)? siz : MAXGAP; i0++; } } else break; } /* reverse the order of jmps */ for (j = 0, i0--; j < i0; j++, i0--) { i = pp[0].n[j]; pp[0].n[j] = pp[0].n[i0]; pp[0].n[i0] = i; i = pp[0].x[j]; pp[0].x[j] = pp[0].x[i0]; pp[0].x[i0] = i; } for (j = 0, i1--; j < i1; j++, i1--) { i = pp[1].n[j]; pp[1].n[j] = pp[1].n[i1]; pp[1].n[i1] = i; i = pp[1].x[j]; pp[1].x[j] = pp[1].x[i1]; pp[1].x[i1] = i; } if (fd >= 0) (void) close(fd); if (fj) { (void) unlink(jname); fj = 0; offset = 0; } } /* * write a filled jmp struct offset of the prev one (if any): nw( ) */ writejmps(ix) writejmps int ix; { char *mktemp( ); if (!fj) { if (mktemp(jname) < 0) { fprintf(stderr, "%s: can't mktemp( ) %s\n", prog, jname); cleanup(1); } if ((fj = fopen(jname, "w")) == 0) { fprintf(stderr, "%s: can't write %s\n", prog, jname); exit(1); } } (void) fwrite((char *)&dx[ix].jp, sizeof(struct jmp), 1, fj); (void) fwrite((char *)&dx[ix].offset, sizeof(dx[ix].offset), 1, fj);
TABLE-US-00008 TABLE 2 STOP-1 XXXXXXXXXXXXXXX (Length = 15 amino acids) Comparison Protein XXXXXYYYYYYY (Length = 12 amino acids) % amino acid sequence identity = (the number of identically matching amino acid residues between the two polypeptide sequences as determined by ALIGN-2) divided by (the total number of amino acid residues of the STOP-1 polypeptide) = 5 divided by 15 = 33.3%
TABLE-US-00009 TABLE 3 STOP-1 XXXXXXXXXX (Length = 10 amino acids) Comparison Protein XXXXXYYYYYYZZYZ (Length = 15 amino acids) % amino acid sequence identity = (the number of identically matching amino acid residues between the two polypeptide sequences as determined by ALIGN-2) divided by (the total number of amino acid residues of the STOP-1 polypeptide) = 5 divided by 10 = 50%
TABLE-US-00010 TABLE 4 STOP-1-DNA NNNNNNNNNNNNNN (Length = 14 nucleotides) Comparison DNA NNNNNNLLLLLLLLLL (Length = 16 nucleotides) % nucleic acid sequence identity = (the number of identically matching nucleotides between the two nucleic acid sequences as determined by ALIGN-2) divided by (the total number of nucleotides of the PRO-DNA nucleic acid sequence) = 6 divided by 14 = 42.9%
TABLE-US-00011 TABLE 5 STOP-1-DNA NNNNNNNNNNNN (Length = 12 nucleotides) Comparison DNA NNNNLLLVV (Length = 9 nucleotides) % nucleic acid sequence identity = (the number of identically matching nucleotides between the two nucleic acid sequences as determined by ALIGN-2) divided by (the total number of nucleotides of the PRO-DNA nucleic acid sequence) = 4 divided by 12 = 33.3%
[0209] Compositions and Methods of the Invention
[0210] STOP-1 Polypeptide Variants
[0211] In addition to the full-length native sequence STOP-1 polypeptides described herein, it is contemplated that STOP-1 polypeptide variants can be prepared. STOP-1 polypeptide variants can be prepared by introducing appropriate nucleotide changes into the STOP-1 DNA, and/or by synthesis of the desired STOP-1 polypeptide. Those skilled in the art will appreciate that amino acid changes can alter post-translational processes of the STOP-1 polypeptide such as changing the number or position of glycosylation sites or altering the membrane anchoring characteristics.
[0212] Variations in the native full-length sequence STOP-1 polypeptide or in various domains of the STOP-1 polypeptide described herein, can be made, for example, using any of the techniques and guidelines for conservative and non-conservative mutations set forth, for instance, in U.S. Pat. No. 5,364,934. Variations can be a substitution, deletion or insertion of one or more codons encoding the STOP-1 polypeptide that results in a change in the amino acid sequence of the STOP-1 polypeptide as compared with the native sequence STOP-1 polypeptide. Optionally the variation is by substitution of at least one amino acid with any other amino acid in one or more of the domains of the STOP-1 polypeptide. Guidance in determining which amino acid residue can be inserted, substituted or deleted without adversely affecting the desired activity can be found by comparing the sequence of the STOP-1 polypeptide with that of homologous known protein molecules and minimizing the number of amino acid sequence changes made in regions of high homology. Amino acid substitutions can be the result of replacing one amino acid with another amino acid having similar structural and/or chemical properties, such as the replacement of a leucine with a serine, i.e., conservative amino acid replacements. Insertions or deletions can optionally be in the range of about 1 to 5 amino acids. The variation allowed can be determined by systematically making insertions, deletions or substitutions of amino acids in the sequence and testing the resulting variants for activity exhibited by the full-length or mature native sequence.
[0213] In particular embodiments, conservative substitutions of interest are shown in Table 6 under the heading of preferred substitutions. If such substitutions result in a change in biological activity, then more substantial changes, denominated exemplary substitutions in Table 6, or as further described below in reference to amino acid classes, are introduced and the products screened.
TABLE-US-00012 TABLE 6 Original Exemplary Preferred Residue Substitutions Substitutions Ala (A) val; leu; ile val Arg (R) lys; gln; asn lys Asn (N) gln; his; lys; arg gln Asp (D) glu glu Cys (C) ser ser Gln (Q) asn asn Glu (E) asp asp Gly (G) pro; ala ala His (H) asn; gln; lys; arg arg Ile (I) leu; val; met; ala; phe; leu norleucine Leu (L) norleucine; ile; val; ile met; ala; phe Lys (K) arg; gln; asn arg Met (M) leu; phe; ile leu Phe (F) leu; val; ile; ala; tyr leu Pro (P) ala ala Ser (S) thr thr Thr (T) ser ser Trp (W) tyr; phe tyr Tyr (Y) trp; phe; thr; ser phe Val (V) ile; leu; met; phe; leu ala; norleucine
[0214] Substantial modifications in function or immunological identity of the STOP-1 polypeptide are accomplished by selecting substitutions that differ significantly in their effect on maintaining (a) the structure of the polypeptide backbone in the area of the substitution, for example, as a sheet or helical conformation, (b) the charge or hydrophobicity of the molecule at the target site, or (c) the bulk of the side chain. Naturally occurring residues are divided into groups based on common side-chain properties:
(1) hydrophobic: norleucine, met, ala, val, leu, ile; (2) neutral hydrophilic: cys, ser, thr; (3) acidic: asp, glu; (4) basic: asn, gln, his, lys, arg; (5) residues that influence chain orientation: gly, pro; and (6) aromatic: trp, tyr, phe.
[0215] Non-conservative substitutions will entail exchanging a member of one of these classes for another class. Such substituted residues also can be introduced into the conservative substitution sites or, more preferably, into the remaining (non-conserved) sites.
[0216] The variations can be made using methods known in the art such as oligonucleotide-mediated (site-directed) mutagenesis, alanine scanning, and PCR mutagenesis. Site-directed mutagenesis [Carter et al., Nucl. Acids Res., 13:4331 (1986); Zoller et al., Nucl. Acids Res., 10:6487 (1987)], cassette mutagenesis [Wells et al., Gene, 34:315 (1985)], restriction selection mutagenesis [Wells et al., Philos. Trans. R. Soc. London SerA, 317:415 (1986)] or other known techniques can be performed on the cloned DNA to produce the STOP-1 variant DNA.
[0217] Scanning amino acid analysis can also be employed to identify one or more amino acids along a contiguous sequence. Among the preferred scanning amino acids are relatively small, neutral amino acids. Such amino acids include alanine, glycine, serine, and cysteine. Alanine is typically a preferred scanning amino acid among this group because it eliminates the side-chain beyond the beta-carbon and is less likely to alter the main-chain conformation of the variant [Cunningham and Wells, Science, 244: 1081-1085 (1989)]. Alanine is also typically preferred because it is the most common amino acid. Further, it is frequently found in both buried and exposed positions [Creighton, The Proteins, (W.H. Freeman & Co., N.Y.); Chothia, J. Mol. Biol., 150:1 (1976)]. If alanine substitution does not yield adequate amounts of variant, an isoteric amino acid can be used.
Modifications of STOP-1 Polypeptides
[0218] Covalent modifications of STOP-1 polypeptides are included within the scope of this invention. One type of covalent modification includes reacting targeted amino acid residues of a STOP-1 polypeptide with an organic derivatizing agent that is capable of reacting with selected side chains or the N- or C-terminal residues of the STOP-1 polypeptide. Derivatization with bifunctional agents is useful, for instance, for crosslinking the STOP-1 polypeptide to a water-insoluble support matrix or surface for use in the method for purifying anti-STOP-1 antibodies, and vice-versa. Commonly used crosslinking agents include, e.g., 1,1-bis(diazoacetyl)-2-phenylethane, glutaraldehyde, N-hydroxysuccinimide esters, for example, esters with 4-azidosalicylic acid, homobifunctional imidoesters, including disuccinimidyl esters such as 3,3'-dithiobis(succinimidylpropionate), bifunctional maleimides such as bis-N-maleimido-1,8-octane and agents such as methyl-3-[(p-azidophenyl)dithio]propioimidate.
[0219] Other modifications include deamidation of glutaminyl and asparaginyl residues to the corresponding glutamyl and aspartyl residues, respectively, hydroxylation of proline and lysine, phosphorylation of hydroxyl groups of seryl or threonyl residues, methylation of the -amino groups of lysine, arginine, and histidine side chains [T. E. Creighton, Proteins: Structure and Molecular Properties, W.H. Freeman & Co., San Francisco, pp. 79-86 (1983)], acetylation of the N-terminal amine, and amidation of any C-terminal carboxyl group.
[0220] Another type of covalent modification of the STOP-1 polypeptide included within the scope of this invention comprises altering the native glycosylation pattern of the polypeptide. "Altering the native glycosylation pattern" is intended for purposes herein to mean deleting one or more carbohydrate moieties found in the native sequence STOP-1 polypeptide (either by removing the underlying glycosylation site or by deleting the glycosylation by chemical and/or enzymatic means), and/or adding one or more glycosylation sites that are not present in the native sequence STOP-1 polypeptide. In addition, the phrase includes qualitative changes in the glycosylation of the native proteins, involving a change in the nature and proportions of the various carbohydrate moieties present.
[0221] Addition of glycosylation sites to the STOP-1 polypeptide can be accomplished by altering the amino acid sequence. The alteration can be made, for example, by the addition of, or substitution by, one or more serine or threonine residues to the native sequence STOP-1 polypeptide (for O-linked glycosylation sites). The STOP-1 amino acid sequence can optionally be altered through changes at the DNA level, particularly by mutating the DNA encoding the STOP-1 polypeptide at preselected bases such that codons are generated that will translate into the desired amino acids.
[0222] Another means of increasing the number of carbohydrate moieties on the STOP-1 polypeptide is by chemical or enzymatic coupling of glycosides to the polypeptide. Such methods are described in the art, e.g., in WO 87/05330 published 11 Sep. 1987, and in Aplin and Wriston, CRC Crit. Rev. Biochem., pp. 259-306 (1981).
[0223] Removal of carbohydrate moieties present on the STOP-1 polypeptide can be accomplished chemically or enzymatically or by mutational substitution of codons encoding for amino acid residues that serve as targets for glycosylation. Chemical deglycosylation techniques are known in the art and described, for instance, by Hakimuddin, et al., Arch. Biochem. Biophys., 259:52 (1987) and by Edge et al., Anal. Biochem., 118:131 (1981). Enzymatic cleavage of carbohydrate moieties on polypeptides can be achieved by the use of a variety of endo- and exo-glycosidases as described by Thotakura et al., Meth. Enzymol., 138:350 (1987).
[0224] Another type of covalent modification of the STOP-1 polypeptide comprises linking the STOP-1 polypeptide to one of a variety of nonproteinaceous polymers, e.g., polyethylene glycol (PEG), polypropylene glycol, or polyoxyalkylenes, in the manner set forth in U.S. Pat. Nos. 4,640,835; 4,496,689; 4,301,144; 4,670,417; 4,791,192 or 4,179,337.
[0225] The STOP-1 polypeptide of the present invention can also be modified in a way to form a chimeric molecule comprising the STOP-1 polypeptide fused to another, heterologous polypeptide or amino acid sequence.
[0226] In one embodiment, such a chimeric molecule comprises a fusion of the STOP-1 polypeptide with a protein transduction domain which targets the STOP-1 polypeptide for delivery to various tissues and more particularly across the brain blood barrier, using, for example, the protein transduction domain of human immunodeficiency virus TAT protein (Schwarze et al., 1999, Science 285: 1569-72).
[0227] In another embodiment, such a chimeric molecule comprises a fusion of the STOP-1 polypeptide with a tag polypeptide which provides an epitope to which an anti-tag antibody can selectively bind. The epitope tag is generally placed at the amino- or carboxyl-terminus of the STOP-1 polypeptide. The presence of such epitope-tagged forms of the STOP-1 polypeptide can be detected using an antibody against the tag polypeptide. Also, provision of the epitope tag enables the STOP-1 polypeptide to be readily purified by affinity purification using an anti-tag antibody or another type of affinity matrix that binds to the epitope tag. Various tag polypeptides and their respective antibodies are known in the art. Examples include poly-histidine (poly-His) or poly-histidine-glycine (poly-His-gly) tags; the flu HA tag polypeptide and its antibody 12CA5 [Field et al., Mol. Cell. Biol., 8:2159-2165 (1988)]; the c-myc tag and the 8F9, 3C7, 6E10, G4, B7 and 9E10 antibodies thereto [Evan et al., Molecular and Cellular Biology, 5:3610-3616 (1985)]; and the Herpes Simplex virus glycoprotein D (gD) tag and its antibody [Paborsky et al., Protein Engineering, 3(6):547-553 (1990)]. Other tag polypeptides include the Flag-peptide [Hopp et al., BioTechnology, 6:1204-1210 (1988)]; the KT3 epitope peptide [Martin et al., Science, 255:192-194 (1992)]; an -tubulin epitope peptide [Skinner et al., J. Biol. Chem., 266:15163-15166 (1991)]; and the T7 gene 10 protein peptide tag [Lutz-Freyermuth et al., Proc. Natl. Acad. Sci. USA, 87:6393-6397 (1990)].
[0228] In an alternative embodiment, the chimeric molecule can comprise a fusion of the STOP-1 polypeptide with an immunoglobulin or a particular region of an immunoglobulin. For a bivalent form of the chimeric molecule (also referred to as an "immunoadhesin"), such a fusion could be to the Fc region of an IgG molecule. Ig fusions of this invention include polypeptides that comprise approximately or only residues 94-243, residues 33-53 or residues 33-52 of human STOP-1 in place of at least one variable region within an Ig molecule. In a particularly preferred embodiment, the immunoglobulin fusion includes the hinge, CH2 and CH3, or the hinge, CH1, CH2 and CH3 regions of an IgG1 molecule. For the production of immunoglobulin fusions see also, U.S. Pat. No. 5,428,130 issued Jun. 27, 1995.
Preparation of the STOP-1 Polypeptide
[0229] The description below relates primarily to production of STOP-1 polypeptides by culturing cells transformed or transfected with a vector containing nucleic acid encoding STOP-1 polypeptides. It is, of course, contemplated that alternative methods that are known in the art can be employed to prepare the STOP-1 polypeptide. For instance, the STOP-1 polypeptide sequence, or portions thereof, can be produced by direct peptide synthesis using solid-phase techniques. See, e.g., Stewart et al., Solid-Phase Peptide Synthesis (W.H. Freeman Co.: San Francisco, Calif., 1969); Merrifield, J. Am. Chem. Soc., 85: 2149-2154 (1963). In vitro protein synthesis can be performed using manual techniques or by automation. Automated synthesis can be accomplished, for instance, with an Applied Biosystems Peptide Synthesizer (Foster City, Calif.) using manufacturer's instructions. Various portions of the STOP-1 polypeptide can be chemically synthesized separately and combined using chemical or enzymatic methods to produce the full-length STOP-1 polypeptide.
Selection and Transformation of Host Cells
[0230] Host cells are transfected or transformed with expression or cloning vectors described herein for STOP-1 polypeptide production and cultured in conventional nutrient media modified as appropriate for inducing promoters, selecting transformants, or amplifying the genes encoding the desired sequences. The culture conditions, such as media, temperature, pH, and the like, can be selected by the skilled artisan without undue experimentation. In general, principles, protocols, and practical techniques for maximizing the productivity of cell cultures can be found in Mammalian Cell Biotechnology: A Practical Approach, M. Butler, ed. (IRL Press, 1991) and Sambrook et al., supra.
[0231] Methods of transfection are known to the ordinarily skilled artisan, for example, CaPO4 treatment and electroporation. Depending on the host cell used, transformation is performed using standard techniques appropriate to such cells. The calcium treatment employing calcium chloride, as described in Sambrook et al., supra, or electroporation is generally used for prokaryotes or other cells that contain substantial cell-wall barriers. Infection with Agrobacterium tumefaciens is used for transformation of certain plant cells, as described by Shaw et al., Gene, 23: 315 (1983) and WO 89/05859 published 29 Jun. 1989. For mammalian cells without such cell walls, the calcium phosphate precipitation method of Graham and van der Eb, Virology, 52:456-457 (1978) can be employed. General aspects of mammalian cell host system transformations have been described in U.S. Pat. No. 4,399,216. Transformations into yeast are typically carried out according to the method of Van Solingen et al., J. Bact., 130: 946 (1977) and Hsiao et al., Proc. Natl. Acad. Sci. (USA), 76: 3829 (1979). However, other methods for introducing DNA into cells, such as by nuclear microinjection, electroporation, bacterial protoplast fusion with intact cells, or polycations, e.g., polybrene or polyornithine, can also be used. For various techniques for transforming mammalian cells, see, Keown et al., Methods in Enzymology, 185: 527-537 (1990) and Mansour et al., Nature, 336: 348-352 (1988).
[0232] Suitable host cells for cloning or expressing the DNA in the vectors herein include prokaryote, yeast, or higher eukaryote cells. Suitable prokaryotes include, but are not limited to, eubacteria, such as Gram-negative or Gram-positive organisms, for example, Enterobacteriaceae such as E. coli. Various E. coli strains are publicly available, such as E. coli K12 strain MM294 (ATCC 31,446); E. coli X1776 (ATCC 31,537); E. coli strain W3110 (ATCC 27,325); and K5 772 (ATCC 53,635). Other suitable prokaryotic host cells include Enterobacteriaceae such as Escherichia, e.g., E. coli, Enterobacter, Erwinia, Klebsiella, Proteus, Salmonella, e.g., Salmonella typhimurium, Serratia, e.g., Serratia marcescans, and Shigella, as well as Bacilli such as B. subtilis and B. licheniformis (e.g., B. licheniformis 41P disclosed in DD 266,710 published 12 Apr. 1989), Pseudomonas such as P. aeruginosa, and Streptomyces. These examples are illustrative rather than limiting. Strain W3110 is one particularly preferred host or parent host because it is a common host strain for recombinant DNA product fermentations. Preferably, the host cell secretes minimal amounts of proteolytic enzymes. For example, strain W3110 can be modified to effect a genetic mutation in the genes encoding proteins endogenous to the host, with examples of such hosts including E. coli W3110 strain 1A2, which has the complete genotype tonA; E. coli W3110 strain 9E4, which has the complete genotype tonA ptr3; E. coli W3110 strain 27C7 (ATCC 55,244), which has the complete genotype tonA ptr3 phoA E15 (argF-lac)169 degP ompT kanr; E. coli W3110 strain 37D6, which has the complete genotype tonA ptr3 phoA E15 (argF-lac)169 degP ompT rbs7 ilvG kanr; E. coli W3110 strain 40B4, which is strain 37D6 with a non-kanamycin resistant degP deletion mutation; and an E. coli strain having mutant periplasmic protease disclosed in U.S. Pat. No. 4,946,783 issued 7 Aug. 1990. Alternatively, in vitro methods of cloning, e.g., PCR or other nucleic acid polymerase reactions, are suitable.
[0233] In addition to prokaryotes, eukaryotic microbes such as filamentous fungi or yeast are suitable cloning or expression hosts for vectors encoding the STOP-1 polypeptide. Saccharomyces cerevisiae is a commonly used lower eukaryotic host microorganism. Others include Schizosaccharomyces pombe (Beach and Nurse, Nature, 290: 140 [1981]; EP 139,383 published 2 May 1985); Kluyveromyces hosts (U.S. Pat. No. 4,943,529; Fleer et al., Bio/Technology, 9: 968-975 (1991)) such as, e.g., K. lactis (MW98-8C, CBS683, CBS4574; Louvencourt et al., J. Bacteriol., 737 [1983]), K. fragilis (ATCC 12,424), K. bulgaricus (ATCC 16,045), K. wickeramii (ATCC 24,178), K. waltii (ATCC 56,500), K. drosophilarum (ATCC 36,906; Van den Berg et al., Bio/Technology, 8: 135 (1990)), K. thermotolerans, and K. marxianus; yarrowia (EP 402,226); Pichia pastoris (EP 183,070; Sreekrishna et al., J. Basic Microbiol., 28: 265-278 [1988]); Candida; Trichoderma reesia (EP 244,234); Neurospora crassa (Case et al., Proc. Natl. Acad. Sci. USA, 76: 5259-5263 [1979]); Schwanniomyces such as Schwanniomyces occidentalis (EP 394,538 published 31 Oct. 1990); and filamentous fungi such as, e.g., Neurospora, Penicillium, Tolypocladium (WO 91/00357 published 10 Jan. 1991), and Aspergillus hosts such as A. nidulans (Ballance et al., Biochem. Biophys. Res. Commun., 112: 284-289 [1983]; Tilburn et al., Gene, 26: 205-221 [1983]; Yelton et al., Proc. Natl. Acad. Sci. USA, 81: 1470-1474 [1984]) and A. niger (Kelly and Hynes, EMBO J., 4: 475-479 [1985]). Methylotropic yeasts are suitable herein and include, but are not limited to, yeast capable of growth on methanol selected from the genera consisting of Hansenula, Candida, Kloeckera, Pichia, Saccharomyces, Torulopsis, and Rhodotorula. A list of specific species that are exemplary of this class of yeasts can be found in C. Anthony, The Biochemistry of Methylotrophs, 269 (1982).
[0234] Suitable host cells for the expression of nucleic acid encoding glycosylated STOP-1 polypeptides are derived from multicellular organisms. Examples of invertebrate cells include insect cells such as Drosophila S2 and Spodoptera Sf9, as well as plant cells. Examples of useful mammalian host cell lines include Chinese hamster ovary (CHO) and COS cells. More specific examples include monkey kidney CV1 line transformed by SV40 (COS-7, ATCC CRL 1651); human embryonic kidney line (293 or 293 cells subcloned for growth in suspension culture, Graham et al., J. Gen. Virol., 36: 59 (1977)); Chinese hamster ovary cells/-DHFR(CHO, Urlaub and Chasin, Proc. Natl. Acad. Sci. USA, 77:4216 (1980)); mouse sertoli cells (TM4, Mather, Biol. Reprod., 23:243-251 (1980)); human lung cells (W138, ATCC CCL 75); human liver cells (Hep G2, HB 8065); and mouse mammary tumor (MMT 060562, ATCC CCL51). The selection of the appropriate host cell is deemed to be within the skill in the art.
Selection and Use of a Replicable Vector
[0235] The nucleic acid (e.g., cDNA or genomic DNA) encoding a polypeptide or antibody of this invention can be inserted into a replicable vector for cloning (amplification of the DNA) or for expression. Various vectors are publicly available. The vector can, for example, be in the form of a plasmid, cosmid, viral particle, or phage. The appropriate nucleic acid sequence can be inserted into the vector by a variety of procedures. In general, DNA is inserted into an appropriate restriction endonuclease site(s) using techniques known in the art. Vector components generally include, but are not limited to, one or more of a signal sequence if the sequence is to be secreted, an origin of replication, one or more marker genes, an enhancer element, a promoter, and a transcription termination sequence. Construction of suitable vectors containing one or more of these components employs standard ligation techniques that are known to the skilled artisan.
[0236] The polypeptide or antibody of this invention can be produced recombinantly not only directly, but also as a fusion polypeptide with a heterologous polypeptide, which can be a signal sequence or other polypeptide having a specific cleavage site at the N-terminus of the mature protein or polypeptide. In general, the signal sequence can be a component of the vector, or it can be a part of the DNA encoding the polypeptide or antibody that is inserted into the vector. The signal sequence can be a prokaryotic signal sequence selected, for example, from the group of the alkaline phosphatase, penicillinase, 1 pp, or heat-stable enterotoxin II leaders. For yeast secretion the signal sequence can be, e.g., the yeast invertase leader, alpha factor leader (including Saccharomyces and Kluyveromyces-factor leaders, the latter described in U.S. Pat. No. 5,010,182), or acid phosphatase leader, the C. albicans glucoamylase leader (EP 362,179 published 4 Apr. 1990), or the signal described in WO 90/13646 published 15 Nov. 1990. In mammalian cell expression, mammalian signal sequences can be used to direct secretion of the protein, such as signal sequences from secreted polypeptides of the same or related species, as well as viral secretory leaders.
[0237] Both expression and cloning vectors contain a nucleic acid sequence that enables the vector to replicate in one or more selected host cells. Such sequences are known for a variety of bacteria, yeast, and viruses. The origin of replication from the plasmid pBR322 is suitable for most Gram-negative bacteria, the 2 u plasmid origin is suitable for yeast, and various viral origins (SV40, polyoma, adenovirus, VSV, or BPV) are useful for cloning vectors in mammalian cells.
[0238] Expression and cloning vectors will typically contain a selection gene, also termed a selectable marker. Typical selection genes encode proteins that (a) confer resistance to antibiotics or other toxins, e.g., ampicillin, neomycin, methotrexate, or tetracycline, (b) complement auxotrophic deficiencies, or (c) supply critical nutrients not available from complex media, e.g., the gene encoding D-alanine racemase for Bacilli.
[0239] An example of suitable selectable markers for mammalian cells are those that enable the identification of cells competent to take up the nucleic acid encoding the polypeptide or antibody such as DHFR or thymidine kinase. An appropriate host cell when wild-type DHFR is employed is the CHO cell line deficient in DHFR activity, prepared and propagated as described by Urlaub et al., Proc. Natl. Acad. Sci. USA, 77: 4216 (1980). A suitable selection gene for use in yeast is the trp1 gene present in the yeast plasmid YRp7. Stinchcomb et al., Nature, 282: 39 (1979); Kingsman et al., Gene, 7: 141 (1979); Tschemper et al., Gene, 10: 157 (1980). The trp1 gene provides a selection marker for a mutant strain of yeast lacking the ability to grow in tryptophan, for example, ATCC No. 44076 or PEP4-1. Jones, Genetics, 85: 12 (1977).
[0240] Expression and cloning vectors usually contain a promoter operably linked to the nucleic acid sequence encoding the polypeptide or antibody of this invention to direct mRNA synthesis. Promoters recognized by a variety of potential host cells are known. Promoters suitable for use with prokaryotic hosts include the -lactamase and lactose promoter systems (Chang et al., Nature, 275: 615 (1978); Goeddel et al., Nature, 281: 544 (1979)), alkaline phosphatase, a tryptophan (trp) promoter system (Goeddel, Nucleic Acids Res., 8: 4057 (1980); EP 36,776), and hybrid promoters such as the tac promoter (deBoer et al., Proc. Natl. Acad. Sci. USA, 80: 21-25 (1983)). Promoters for use in bacterial systems also will contain a Shine-Dalgarno (S.D.) sequence operably linked to the DNA encoding the polypeptide or antibody of this invention.
[0241] Examples of suitable promoting sequences for use with yeast hosts include the promoters for 3-phosphoglycerate kinase (Hitzeman et al., J. Biol. Chem., 255: 2073 (1980)) or other glycolytic enzymes (Hess et al., J. Adv. Enzyme Reg., 7: 149 (1968); Holland, Biochemistry, 17: 4900 (1978)), such as enolase, glyceraldehyde-3-phosphate dehydrogenase, hexokinase, pyruvate decarboxylase, phosphofructokinase, glucose-6-phosphate isomerase, 3-phosphoglycerate mutase, pyruvate kinase, triosephosphate isomerase, phosphoglucose isomerase, and glucokinase.
[0242] Other yeast promoters that are inducible promoters having the additional advantage of transcription controlled by growth conditions are the promoter regions for alcohol dehydrogenase 2, isocytochrome C, acid phosphatase, degradative enzymes associated with nitrogen metabolism, metallothionein, glyceraldehyde-3-phosphate dehydrogenase, and enzymes responsible for maltose and galactose utilization. Suitable vectors and promoters for use in yeast expression are further described in EP 73,657.
[0243] Nucleic acid transcription from vectors in mammalian host cells is controlled, for example, by promoters obtained from the genomes of viruses such as polyoma virus, fowlpox virus (UK 2,211,504 published 5 Jul. 1989), adenovirus (such as Adenovirus 2), bovine papilloma virus, avian sarcoma virus, cytomegalovirus, a retrovirus, hepatitis-B virus, and Simian Virus 40 (SV40); by heterologous mammalian promoters, e.g., the actin promoter or an immunoglobulin promoter; and by heat-shock promoters, provided such promoters are compatible with the host cell systems.
[0244] Transcription of a DNA encoding a polypeptide or antibody of this invention by higher eukaryotes can be increased by inserting an enhancer sequence into the vector. Enhancers are cis-acting elements of DNA, usually about from 10 to 300 bp, that act on a promoter to increase its transcription. Many enhancer sequences are now known from mammalian genes (globin, elastase, albumin, -fetoprotein, and insulin). Typically, however, one will use an enhancer from a eukaryotic cell virus. Examples include the SV40 enhancer on the late side of the replication origin (bp 100-270), the cytomegalovirus early promoter enhancer, the polyoma enhancer on the late side of the replication origin, and adenovirus enhancers. The enhancer can be spliced into the vector at a position 5' or 3' to the sequence coding for a polypeptide or antibody of this invention, but is preferably located at a site 5' from the promoter.
[0245] Expression vectors used in eukaryotic host cells (yeast, fungi, insect, plant, animal, human, or nucleated cells from other multicellular organisms) will also contain sequences necessary for the termination of transcription and for stabilizing the mRNA. Such sequences are commonly available from the 5' and, occasionally 3', untranslated regions of eukaryotic or viral DNAs or cDNAs. These regions contain nucleotide segments transcribed as polyadenylated fragments in the untranslated portion of the mRNA encoding polypeptide or antibody.
[0246] Still other methods, vectors, and host cells suitable for adaptation to the synthesis of the polypeptide or antibody of this invention in recombinant vertebrate cell culture are described in Gething et al., Nature, 293: 620-625 (1981); Mantei et al., Nature, 281: 40-46 (1979); EP 117,060; and EP 117,058.
Detecting Gene Amplification/Expression
[0247] Gene amplification and/or expression can be measured in a sample directly, for example, by conventional Southern blotting, Northern blotting to quantitate the transcription of mRNA (Thomas, Proc. Natl. Acad. Sci. USA, 77:5201-5205 (1980)), dot blotting (DNA analysis), or in situ hybridization, using an appropriately labeled probe, based on the sequences provided herein. Alternatively, antibodies can be employed that can recognize specific duplexes, including DNA duplexes, RNA duplexes, and DNA-RNA hybrid duplexes or DNA-protein duplexes. The antibodies in turn can be labeled and the assay can be carried out where the duplex is bound to a surface, so that upon the formation of duplex on the surface, the presence of antibody bound to the duplex can be detected.
[0248] Gene expression, alternatively, can be measured by immunological methods, such as immunohistochemical staining of cells or tissue sections and assay of cell culture or body fluids, to quantitate directly the expression of gene product. Antibodies useful for immunohistochemical staining and/or assay of sample fluids can be either monoclonal or polyclonal, and can be prepared in any mammal or can be synthesized (e.g., the monoclonal antibodies of this invention). Conveniently, the antibodies can be prepared against a native-sequence STOP-1 polypeptide or against a synthetic peptide based on the DNA sequences provided herein or against exogenous sequence fused to DNA encoding the STOP-1 polypeptide and encoding a specific antibody epitope.
Purification of STOP-1 Polypeptides
[0249] Forms of STOP-1 polypeptides can be recovered from culture medium or from host cell lysates. If membrane-bound, it can be released from the membrane using a suitable detergent solution (e.g., TRITON-X® 100) or by enzymatic cleavage. Cells employed in expression of nucleic acid encoding the STOP-1 polypeptide can be disrupted by various physical or chemical means, such as freeze-thaw cycling, sonication, mechanical disruption, or cell-lysing agents. According to one embodiment, it is desireabel that the STOP-1 polypeptide is purified from recombinant cell proteins or polypeptides. The following procedures are exemplary of suitable purification procedures: by fractionation on an ion-exchange column; ethanol precipitation; reverse phase HPLC; chromatography on silica or on a cation-exchange resin such as DEAE; chromatofocusing; SDS-PAGE; ammonium sulfate precipitation; gel filtration using, for example, Sephadex G-75; protein A Sepharose columns to remove contaminants such as IgG; and metal chelating columns to bind epitope-tagged forms of the STOP-1 polypeptide. Various methods of protein purification can be employed and such methods are known in the art and described, for example, in Deutscher, Methods in Enzymology, 182 (1990); Scopes, Protein Purification: Principles and Practice (Springer-Verlag: New York, 1982). The purification step(s) selected will depend, for example, on the nature of the production process used and the particular STOP-1 polypeptide produced. According to one embodiment, the STOP-1 polypeptides are purified by affinity chromatography using an antibody of this invention.
Assaying Inhibition of Cell Proliferation
[0250] The inhibitory activity of antagonists of this invention can be measured using the assays of Examples 13-14 below and other assays known in the art.
[0251] Animal models of tumors and cancers (e.g., breast cancer, colon cancer, prostate cancer, lung cancer, etc.) include both non-recombinant and recombinant (transgenic) animals. Non-recombinant animal models include, for example, rodent, e.g., murine models. Such models can be generated by introducing tumor cells into syngeneic mice using standard techniques, e.g., subcutaneous injection, tail vein injection, spleen implantation, intraperitoneal implantation, implantation under the renal capsule, or orthopin implantation, e.g., colon cancer cells implanted in colonic tissue. See, e.g., PCT publication No. WO 97/33551, published Sep. 18, 1997. Probably the most often used animal species in oncological studies are immunodeficient mice and, in particular, nude mice. The observation that the nude mouse with thymic hypo/aplasia could successfully act as a host for human tumor xenografts has lead to its widespread use for this purpose. The autosomal recessive nu gene has been introduced into a very large number of distinct congenic strains of nude mouse, including, for example, ASW, A/He, AKR, BALB/c, B10.LP, C17, C3H, C57BL, C57, CBA, DBA, DDD, I/st, NC, NFR, NFS, NFS/N, NZB, NZC, NZW, P, RIII, and SJL. In addition, a wide variety of other animals with inherited immunological defects other than the nude mouse have been bred and used as recipients of tumor xenografts. For further details see, e.g., The Nude Mouse in Oncology Research, E. Boven and B. Winograd, eds. (CRC Press, Inc., 1991).
[0252] The cells introduced into such animals can be derived from known tumor/cancer cell lines, such as any of the above-listed tumor cell lines, and, for example, the B104-1-1 cell line (stable NIH-3T3 cell line transfected with the neu protooncogene); ras-transfected NIH-3T3 cells; Caco-2 (ATCC HTB-37); or a moderately well-differentiated grade II human colon adenocarcinoma cell line, HT-29 (ATCC HTB-38); or from tumors and cancers. Samples of tumor or cancer cells can be obtained from patients undergoing surgery, using standard conditions involving freezing and storing in liquid nitrogen. Karmali et al., Br. J. Cancer, 48: 689-696 (1983).
[0253] Tumor cells can be introduced into animals such as nude mice by a variety of procedures. The subcutaneous (s.c.) space in mice is very suitable for tumor implantation. Tumors can be transplanted s.c. as solid blocks, as needle biopsies by use of a trochar, or as cell suspensions. For solid-block or trochar implantation, tumor tissue fragments of suitable size are introduced into the s.c. space. Cell suspensions are freshly prepared from primary tumors or stable tumor cell lines, and injected subcutaneously. Tumor cells can also be injected as subdermal implants. In this location, the inoculum is deposited between the lower part of the dermal connective tissue and the s.c. tissue.
[0254] Animal models of breast cancer can be generated, for example, by implanting rat neuroblastoma cells (from which the neu oncogene was initially isolated), or neu-transformed NIH-3T3 cells into nude mice, essentially as described by Drebin et al. Proc. Nat. Acad. Sci. USA, 83: 9129-9133 (1986).
[0255] Similarly, animal models of colon cancer can be generated by passaging colon cancer cells in animals, e.g., nude mice, leading to the appearance of tumors in these animals. An orthotopic transplant model of human colon cancer in nude mice has been described, for example, by Wang et al., Cancer Research, 54: 4726-4728 (1994) and Too et al., Cancer Research, 55: 681-684 (1995). This model is based on the so-called "METAMOUSE®" sold by AntiCancer, Inc., (San Diego, Calif.).
[0256] Tumors that arise in animals can be removed and cultured in vitro. Cells from the in vitro cultures can then be passaged to animals. Such tumors can serve as targets for further testing or drug screening. Alternatively, the tumors resulting from the passage can be isolated and RNA from pre-passage cells and cells isolated after one or more rounds of passage analyzed for differential expression of genes of interest. Such passaging techniques can be performed with any known tumor or cancer cell lines.
[0257] For example, Meth A, CMS4, CMS5, CMS21, and WEHI-164 are chemically induced fibrosarcomas of BALB/c female mice (DeLeo et al., J. Exp. Med., 146: 720 (1977)), which provide a highly controllable model system for studying the anti-tumor activities of various agents. Palladino et al., J. Immunol., 138: 4023-4032 (1987). Briefly, tumor cells are propagated in vitro in cell culture. Prior to injection into the animals, the cell lines are washed and suspended in buffer, at a cell density of about 10×106 to 10×107 cells/ml. The animals are then infected subcutaneously with 10 to 100 ul of the cell suspension, allowing one to three weeks for a tumor to appear.
[0258] In addition, the Lewis lung (3LL) carcinoma of mice, which is one of the most thoroughly studied experimental tumors, can be used as an investigational tumor model. Efficacy in this tumor model has been correlated with beneficial effects in the treatment of human patients diagnosed with small-cell carcinoma of the lung (SCCL). This tumor can be introduced in normal mice upon injection of tumor fragments from an affected mouse or of cells maintained in culture. Zupi et al., Br. J. Cancer, 41: suppl. 4, 30 (1980). Evidence indicates that tumors can be started from injection of even a single cell and that a very high proportion of infected tumor cells survive. For further information about this tumor model see, Zacharski, Haemostasis, 16: 300-320 (1986).
[0259] One way of evaluating the efficacy of a test compound in an animal model with an implanted tumor is to measure the size of the tumor before and after treatment. Traditionally, the size of implanted tumors has been measured with a slide caliper in two or three dimensions. The measure limited to two dimensions does not accurately reflect the size of the tumor; therefore, it is usually converted into the corresponding volume by using a mathematical formula. However, the measurement of tumor size is very inaccurate. The therapeutic effects of a drug candidate can be better described as treatment-induced growth delay and specific growth delay. Another important variable in the description of tumor growth is the tumor volume doubling time. Computer programs for the calculation and description of tumor growth are also available, such as the program reported by Rygaard and Spang-Thomsen, Proc. 6th Int. Workshop on Immune-Deficient Animals, Wu and Sheng eds. (Basel, 1989), p. 301. It is noted, however, that necrosis and inflammatory responses following treatment can actually result in an increase in tumor size, at least initially. Therefore, these changes need to be carefully monitored, by a combination of a morphometric method and flow cytometric analysis.
[0260] Further, recombinant (transgenic) animal models can be engineered by introducing the coding portion of the STOP-1 gene identified herein into the genome of animals of interest, using standard techniques for producing transgenic animals. Animals that can serve as a target for transgenic manipulation include, without limitation, mice, rats, rabbits, guinea pigs, sheep, goats, pigs, and non-human primates, e.g., baboons, chimpanzees and monkeys. Techniques known in the art to introduce a transgene into such animals include pronucleic microinjection (U.S. Pat. No. 4,873,191); retrovirus-mediated gene transfer into germ lines (e.g., Van der Putten et al., Proc. Natl. Acad. Sci. USA, 82: 6148-615 (1985)); gene targeting in embryonic stem cells (Thompson et al., Cell, 56: 313-321 (1989)); electroporation of embryos (Lo, Mol. Cell. Biol., 3: 1803-1814 (1983)); and sperm-mediated gene transfer. Lavitrano et al., Cell, 57: 717-73 (1989). For a review, see for example, U.S. Pat. No. 4,736,866.
[0261] For the purpose of the present invention, transgenic animals include those that carry the transgene only in part of their cells ("mosaic animals"). The transgene can be integrated either as a single transgene, or in concatamers, e.g., head-to-head or head-to-tail tandems. Selective introduction of a transgene into a particular cell type is also possible by following, for example, the technique of Lasko et al., Proc. Natl. Acad. Sci. USA, 89: 6232-636 (1992). The expression of the transgene in transgenic animals can be monitored by standard techniques. For example, Southern blot analysis or PCR amplification can be used to verify the integration of the transgene. The level of mRNA expression can then be analyzed using techniques such as in situ hybridization, Northern blot analysis, PCR, or immunocytochemistry. The animals are further examined for signs of tumor or cancer development.
[0262] Alternatively, "knock-out" animals can be constructed that have a defective or altered gene encoding a STOP-1 polypeptide identified herein, as a result of homologous recombination between the endogenous gene encoding the STOP-1 polypeptide and altered genomic DNA encoding the same polypeptide introduced into an embryonic cell of the animal. For example, cDNA encoding a particular STOP-1 polypeptide can be used to clone genomic DNA encoding that polypeptide in accordance with established techniques. A portion of the genomic DNA encoding a particular STOP-1 polypeptide can be deleted or replaced with another gene, such as a gene encoding a selectable marker that can be used to monitor integration. Typically, several kilobases of unaltered flanking DNA (both at the 5' and 3' ends) are included in the vector. See, e.g., Thomas and Capecchi, Cell, 51: 503 (1987) for a description of homologous recombination vectors. The vector is introduced into an embryonic stem cell line (e.g., by electroporation) and cells in which the introduced DNA has homologously recombined with the endogenous DNA are selected. See, e.g., Li et al., Cell, 69: 915 (1992). The selected cells are then injected into a blastocyst of an animal (e.g., a mouse or rat) to form aggregation chimeras. See, e.g., Bradley, in Teratocarcinomas and Embryonic Stem Cells: A Practical Approach, E. J. Robertson, ed. (IRL: Oxford, 1987), pp. 113-152. A chimeric embryo can then be implanted into a suitable pseudopregnant female foster animal and the embryo brought to term to create a "knock-out" animal. Progeny harboring the homologously recombined DNA in their germ cells can be identified by standard techniques and used to breed animals in which all cells of the animal contain the homologously recombined DNA. Knockout animals can be characterized, for instance, by their ability to defend against certain pathological conditions and by their development of pathological conditions due to absence of the STOP-1 polypeptide.
[0263] The efficacy of antibodies specifically binding the STOP-1 polypeptides identified herein, and other drug candidates, can be tested also in the treatment of spontaneous animal tumors. A suitable target for such studies is the feline oral squamous cell carcinoma (SCC). Feline oral SCC is a highly invasive, malignant tumor that is the most common oral malignancy of cats, accounting for over 60% of the oral tumors reported in this species. It rarely metastasizes to distant sites, although this low incidence of metastasis can merely be a reflection of the short survival times for cats with this tumor. These tumors are usually not amenable to surgery, primarily because of the anatomy of the feline oral cavity. At present, there is no effective treatment for this tumor. Prior to entry into the study, each cat undergoes complete clinical examination and biopsy, and is scanned by computed tomography (CT). Cats diagnosed with sublingual oral squamous cell tumors are excluded from the study. The tongue can become paralyzed as a result of such tumor, and even if the treatment kills the tumor, the animals may not be able to feed themselves. Each cat is treated repeatedly, over a longer period of time. Photographs of the tumors will be taken daily during the treatment period, and at each subsequent recheck. After treatment, each cat undergoes another CT scan. CT scans and thoracic radiograms are evaluated every 8 weeks thereafter. The data are evaluated for differences in survival, response, and toxicity as compared to control groups. Positive response may require evidence of tumor regression, preferably with improvement of quality of life and/or increased life span.
[0264] In addition, other spontaneous animal tumors, such as fibrosarcoma, adenocarcinoma, lymphoma, chondroma, or leiomyosarcoma of dogs, cats, and baboons can also be tested. Of these, mammary adenocarcinoma in dogs and cats is a preferred model as its appearance and behavior are very similar to those in humans. However, the use of this model is limited by the rare occurrence of this type of tumor in animals.
Assays for Evaluating Angiogenic or Vasculogenic Activity
[0265] Assays that are useful for measuring the pro-angiogenic, antiangiogenic, pro-vasculogenic or anti-vasculogenic activity of the agonists, progenitors, antagonists of this invention include the assays of Examples 14 and 26 or other suitable assays known in the art such as those included below.
[0266] Assays for wound-healing activity include, for example, those described in Winter, Epidermal Wound Healing, Maibach, H I and Rovee, D T, eds. (Year Book Medical Publishers, Inc., Chicago), pp. 71-112, as modified by the article of Eaglstein and Mertz, J. Invest. Dermatol., 71: 382-384 (1978).
[0267] Assays for endothelial cell proliferation include, for example, those described in WO 02/00690 or United States Patent Publication No. 20010036955A1.
[0268] Assays for evaluating inhibition of angiogenesis include, for example, the assay described in United States Patent Publication No. 20010036955A1.
[0269] Assays for measuring inhibition of endothelial tube formation include, for example, the assay described in United States Patent Publication No. 20010036955A1.
Antibody Binding Studies
[0270] Antibody binding studies can be carried out using known assay methods, such as competitive binding assays, direct and indirect sandwich assays, and immunoprecipitation assays. Zola, Monoclonal Antibodies: A Manual of Techniques (CRC Press, Inc., 1987), pp. 147-158.
[0271] Competitive binding assays rely on the ability of a labeled standard to compete with the test sample analyte for binding with a limited amount of antibody. The amount of target protein in the test sample is inversely proportional to the amount of standard that becomes bound to the antibodies. To facilitate determining the amount of standard that becomes bound, the antibodies preferably are insolubilized before or after the competition, so that the standard and analyte that are bound to the antibodies can conveniently be separated from the standard and analyte that remain unbound.
[0272] Sandwich assays involve the use of two antibodies, each capable of binding to a different immunogenic portion, or epitope, of the protein to be detected. In a sandwich assay, the test sample analyte is bound by a first antibody that is immobilized on a solid support, and thereafter a second antibody binds to the analyte, thus forming an insoluble three-part complex. See, e.g., U.S. Pat. No. 4,376,110. The second antibody can itself be labeled with a detectable moiety (direct sandwich assays) or can be measured using an anti-immunoglobulin antibody that is labeled with a detectable moiety (indirect sandwich assay). For example, one type of sandwich assay is an ELISA assay, in which case the detectable moiety is an enzyme.
[0273] Competitive ELISA assays can be performed to screen polypeptides, antibodies or antagonists for those that specifically bind to STOP-1, which binding can be inhibited by a monoclonal antibody of this invention.
[0274] In one example, a competitive ELISA assay can be conducted following the methods described in the Examples (e.g., Example 22). A full length or short form of native STOP-1 protein (2 ug/ml in PBS) can be coated on a microtiter plate at 4° C. overnight or at room temperature for 2 hours. The wells can be blocked by adding 65 ul 1% BSA for 30 minutes followed by 40 ul 1% Tween20 for another 30 minutes. Next, the wells can be washed with PBS--0.05% Tween20 5 times. Various concentrations of S7, S16, S4, F15 or S9 antibody (in ELISA buffer) can be incubated in the wells for 30 minutes at room temperature. Then, polypeptides or antibodies to be tested can be added to different wells for 10 minutes at a concentration that would normally produce 90% binding capacity in the absence of the S7, S16, S4, F15 or S9 antibody. Next, the wells can be washed with PBS--0.05% Tween20 5 times. Binding can be quantified by methods known in the art.
[0275] For immunohistochemistry, the tissue sample can be fresh or frozen or can be embedded in paraffin and fixed with a preservative such as formalin, for example.
Cell-Based Tumor Assays
[0276] Cell-based assays and animal models for proliferative disorders, such as tumors, can be used to verify the inhibitory activity of the antagonists of this invention. Useful cell-based assays, animal models and methods include, for example, those set forth in the Examples below.
[0277] For example, cells of a cell type known to be involved in a proliferative disorder are transfected with STOP-1 cDNAs herein, and the ability of these cDNAs to induce excessive growth or inhibit growth is analyzed in the presence or absence of an antagonist. If the proliferative disorder is cancer, suitable tumor cells include, for example, stable tumor cell lines such as the B104-1-1 cell line (stable NIH-3T3 cell line transfected with the neu protooncogene) and ras-transfected NIH-3T3 cells, which can be transfected with the a STOP-1 sequence and monitored for tumorigenic growth. Such transfected cell lines can then be used to test the ability of poly- or monoclonal antibodies or antibody compositions to inhibit tumorigenic cell growth by exerting cytostatic or cytotoxic activity on the growth of the transformed cells, or by mediating antibody-dependent cellular cytotoxicity (ADCC).
[0278] In addition, primary cultures derived from tumors in transgenic animals (as described above) can be used in the cell-based assays herein, although stable cell lines are preferred. Techniques to derive continuous cell lines from transgenic animals are known in the art. See, e.g., Small et al., Mol. Cell. Biol., 5: 642-648 (1985).
Gene Therapy
[0279] Described below are methods and compositions whereby disease symptoms can be ameliorated. The STOP-1 polypeptides (including STOP-1 polypeptide variants) described herein, antagonists and antibodies of this invention can be employed in accordance with the present invention by expression of each in vivo, which is often referred to as gene therapy. For example, STOP-1 polypeptide variants can be expressed in cells using these methods. According to one embodiment, the methods or the vectors used to express the STOP-1 polypeptides (including variants) involve the use of a stromal targeting agent to direct the vehicle containing the UNQ polypeptide or nucleic acid molecule to a desired stromal region.
[0280] There are two major approaches to getting the nucleic acid (optionally contained in a vector) into the mammal's cells: in vivo and ex vivo. For in vivo delivery the nucleic acid is injected directly into the mammal, usually at the sites where the STOP-1 polypeptide is required, i.e., the site of synthesis of the STOP-1 polypeptide, if known, and the site (e.g., wound) where biological activity of the STOP-1 polypeptide is needed. For ex vivo treatment, the mammal's cells are removed, the nucleic acid is introduced into these isolated cells, and the modified cells are administered to the mammal either directly or, for example, encapsulated within porous membranes that are implanted into the mammal (see, e.g., U.S. Pat. Nos. 4,892,538 and 5,283,187). There are a variety of techniques available for introducing nucleic acids into viable cells. The techniques vary depending upon whether the nucleic acid is transferred into cultured cells in vitro, or transferred in vivo in the cells of the intended host. Techniques suitable for the transfer of nucleic acid into mammalian cells in vitro include the use of liposomes, electroporation, microinjection, transduction, cell fusion, DEAE-dextran, the calcium phosphate precipitation method, etc. Transduction involves the association of a replication-defective, recombinant viral (preferably retroviral) particle with a cellular receptor, followed by introduction of the nucleic acids contained by the particle into the cell. A commonly used vector for ex vivo delivery of the gene is a retrovirus.
[0281] The currently preferred in vivo nucleic acid transfer techniques include transfection with viral or non-viral vectors (such as adenovirus, lentivirus, Herpes simplex I virus, or adeno-associated virus (AAV)) and lipid-based systems (useful lipids for lipid-mediated transfer of the gene are, for example, DOTMA, DOPE, and DC-Chol; see, e.g., Tonkinson et al., Cancer Investigation, 14(1): 54-65 (1996)). Such vectors are used to synthesize virus that can be used as vehicles for delivering agents, such as antagonists and nucleic acid molecules of this invention. The most preferred vectors for use in gene therapy are viruses, most preferably adenoviruses, AAV, lentiviruses, or retroviruses. A viral vector such as a retroviral vector includes at least one transcriptional promoter/enhancer or locus-defining element(s), or other elements that control gene expression by other means such as alternate splicing, nuclear RNA export, or post-translational modification of messenger. In addition, a viral vector such as a retroviral vector includes a nucleic acid molecule that, when transcribed in the presence of a gene encoding the STOP-1 polypeptide, is operably linked thereto and acts as a translation initiation sequence. Such vector constructs also include a packaging signal, long terminal repeats (LTRs) or portions thereof, and positive and negative strand primer binding sites appropriate to the virus used (if these are not already present in the viral vector). In addition, such vector typically includes a signal sequence for secretion of the STOP-1 polypeptide from a host cell in which it is placed. Preferably the signal sequence for this purpose is a mammalian signal sequence, most preferably the native signal sequence for the STOP-1 polypeptide. Optionally, the vector construct can also include a signal that directs polyadenylation, as well as one or more restriction sites and a translation termination sequence. By way of example, such vectors will typically include a 5' LTR, a tRNA binding site, a packaging signal, an origin of second-strand DNA synthesis, and a 3' LTR or a portion thereof. Other vectors can be used that are non-viral, such as cationic lipids, polylysine, and dendrimers. According to one embodiment, the vehicle has a stromal targeting agent.
[0282] In some situations, it is desirable to provide the nucleic acid source with an agent that targets the target cells, such as an antibody specific for a cell-surface membrane protein or the target cell, a ligand for a receptor on the target cell, etc. Where liposomes are employed, proteins that bind to a cell-surface membrane protein associated with endocytosis can be used for targeting and/or to facilitate uptake, e.g., capsid proteins or fragments thereof tropic for a particular cell type, antibodies for proteins that undergo internalization in cycling, and proteins that target intracellular localization and enhance intracellular half-life. The technique of receptor-mediated endocytosis is described, for example, by Wu et al., J. Biol. Chem., 262: 4429-4432 (1987); and Wagner et al., Proc. Natl. Acad. Sci. USA, 87: 3410-3414 (1990). For a review of the currently known gene marking and gene therapy protocols, see, Anderson et al., Science, 256: 808-813 (1992). See also WO 93/25673 and the references cited therein.
[0283] Suitable gene therapy and methods for making retroviral particles and structural proteins can be found in, e.g., U.S. Pat. No. 5,681,746.
Detecting STOP-1 Mutations
[0284] This invention is also related to the use of the gene encoding the STOP-1 polypeptide as a diagnostic. Detection of a mutated form of the STOP-1 polypeptide can be indicative of a proclivity for developing a proliferative disorder. Detection of levels of the STOP-1 polypeptide in the tissue of a mammal over the levels of the same tissue in a normal mammal can also be indicative of proclivity of developing a proliferative disorder (below).
[0285] Individuals carrying mutations in the genes encoding a human STOP-1 polypeptide can be detected at the DNA level by a variety of techniques. Nucleic acids for diagnosis can be obtained from a mammal's cells, such as from blood, urine, saliva, tissue biopsy, and autopsy material. The genomic DNA can be used directly for detection or can be amplified enzymatically by using PCR (Saiki et al., Nature, 324: 163-166 (1986)) prior to analysis. RNA or cDNA can also be used for the same purpose. As an example, PCR primers complementary to the nucleic acid encoding the STOP-1 polypeptide can be used to identify and analyze the STOP-1 polypeptide mutations. For example, deletions and insertions can be detected by a change in size of the amplified product in comparison to the normal genotype. Point mutations can be identified by hybridizing amplified DNA to radiolabeled RNA encoding the STOP-1 polypeptide, or alternatively, radiolabeled antisense DNA sequences encoding the STOP-1 polypeptide. Perfectly matched sequences can be distinguished from mismatched duplexes by RNase A digestion or by differences in melting temperatures.
[0286] Genetic testing based on DNA sequence differences can be achieved by detection of alteration in electrophoretic mobility of DNA fragments in gels with or without denaturing agents. Small sequence deletions and insertions can be visualized by high resolution gel electrophoresis. DNA fragments of different sequences can be distinguished on denaturing formamidine gradient gels in which the mobilities of different DNA fragments are retarded in the gel at different positions according to their specific melting or partial melting temperatures. See, e.g., Myers et al., Science, 230: 1242 (1985).
[0287] Sequence changes at specific locations may also be revealed by nuclease protection assays, such as RNase and Si protection or the chemical cleavage method, for example, Cotton et al., Proc. Natl. Acad. Sci. USA, 85: 4397-4401 (1985).
[0288] Thus, the detection of a specific DNA sequence can be achieved by methods such as hybridization, RNase protection, chemical cleavage, direct DNA sequencing, or the use of restriction enzymes, e.g., restriction fragment length polymorphisms (RFLP), and Southern blotting of genomic DNA.
[0289] In addition to more conventional gel-electrophoresis and DNA sequencing, mutations in the STOP-1 polypeptide can also be detected by in situ analysis.
Detecting STOP-1 Polypeptide or Nucleic Acid Levels
[0290] Levels of STOP-1 polypeptide or nucleic acid molecules can be detected, e.g., using the reagents disclosed herein in combination with methods known in the art, such as in situ hybridization, RT-PCR, northern blots, western blots, or by using the Examples and reagents provided herein.
[0291] A competition assay can be employed wherein antibodies specific to the STOP-1 polypeptide are attached to a solid support and labeled STOP-1 polypeptide and a sample derived from the host are passed over the solid support and the amount of label detected attached to the solid support can be correlated to a quantity of the STOP-1 polypeptide in the sample.
[0292] In one preferred embodiment, antibodies that specifically bind STOP-1 as described herein are used to monitor STOP-1 protein levels.
Screening Assays for Drug Candidates
[0293] This invention encompasses methods of screening compounds to identify those that mimic the STOP-1 polypeptide activity (agonists) or prevent the effect of the STOP-1 polypeptide (antagonists). Generally, the STOP-1 polypeptide is exposed to the drug candidate by incubation or contact under various conditions. Screening assays for antagonist drug candidates are designed to identify compounds that specifically bind or complex with the native STOP-1 polypeptide. Such screening assays will include assays amenable to high-throughput screening of chemical libraries, making them particularly suitable for identifying small molecule drug candidates.
[0294] The assays can be performed in a variety of formats, including protein-protein binding assays, biochemical screening assays, immunoassays, and cell-based assays in combination with the STOP-1 polypeptide, fragments thereof, or cells expressing the STOP-1 polypeptide or fragments thereof.
[0295] All assays for antagonists are common in that they call for contacting the drug candidate with a STOP-1 polypeptide encoded by a nucleic acid identified herein under conditions and for a time sufficient to allow these two components to interact.
[0296] In binding assays, the interaction is binding and the complex formed can be isolated or detected in the reaction mixture. For example, binding of STOP-1 polypeptide to a cancer cell or an endothelial cells in the absence or presence of the candidate antagonist can be performed in the assays described in the Examples below to evaluate whether the antagonist blocked binding of STOP-1 to the cells. In another embodiment, the STOP-1 polypeptide encoded by the gene identified herein or the drug candidate is immobilized on a solid phase, e.g., on a microtiter plate, by covalent or non-covalent attachments. Non-covalent attachment generally is accomplished by coating the solid surface with a solution of the STOP-1 polypeptide and drying. Alternatively, an immobilized antibody, e.g., a monoclonal antibody, specific for the STOP-1 polypeptide to be immobilized can be used to anchor it to a solid surface. The assay is performed by adding the non-immobilized component, which can be labeled by a detectable label, to the immobilized component, e.g., the coated surface containing the anchored component. When the reaction is complete, the non-reacted components are removed, e.g., by washing, and complexes anchored on the solid surface are detected. When the originally non-immobilized component carries a detectable label, the detection of label immobilized on the surface indicates that complexing occurred. Where the originally non-immobilized component does not carry a label, complexing can be detected, for example, by using a labeled antibody specifically binding the immobilized complex.
[0297] If the candidate compound interacts with but does not bind to a particular STOP-1 polypeptide, its interaction with that polypeptide can be assayed by methods known for detecting protein-protein interactions. Such assays include traditional approaches, such as, e.g., cross-linking, co-immunoprecipitation, and co-purification through gradients or chromatographic columns. In addition, protein-protein interactions can be monitored by using a yeast-based genetic system described by Fields and co-workers (Fields and Song, Nature (London), 340: 245-246 (1989); Chien et al., Proc. Natl. Acad. Sci. USA, 88: 9578-9582 (1991)) as disclosed by Chevray and Nathans, Proc. Natl. Acad. Sci. USA, 89: 5789-5793 (1991). Many transcriptional activators, such as yeast GAL4, consist of two physically discrete modular domains, one acting as the DNA-binding domain, the other one functioning as the transcription-activation domain. The yeast expression system described in the foregoing publications (generally referred to as the "two-hybrid system") takes advantage of this property, and employs two hybrid proteins, one in which the target protein is fused to the DNA-binding domain of GAL4, and another, in which candidate activating proteins are fused to the activation domain. The expression of a GAL1-lacZ reporter gene under control of a GAL4-activated promoter depends on reconstitution of GAL4 activity via protein-protein interaction. Colonies containing interacting polypeptides are detected with a chromogenic substrate for -galactosidase. A complete kit (MATCHMAKER®) for identifying protein-protein interactions between two specific proteins using the two-hybrid technique is commercially available from Clontech. This system can also be extended to map protein domains involved in specific protein interactions as well as to pinpoint amino acid residues that are crucial for these interactions.
[0298] Compounds that interfere with binding between a STOP-1 polypeptide and another protein, including another STOP-1 polypeptide can be tested as follows: usually a reaction mixture is prepared containing the STOP-1 polypeptide and other protein under conditions and for a time allowing for the interaction and binding of the two proteins. To test the ability of a candidate compound to inhibit binding, the reaction is run in the absence and in the presence of the test compound. In addition, a placebo can be added to a third reaction mixture, to serve as positive control. The binding (complex formation) between the test compound and the other polypeptide present in the mixture is monitored as described hereinabove. The formation of a complex in the control reaction(s) but not in the reaction mixture containing the test compound indicates that the test compound interferes with the interaction of the STOP-1 polypeptide and the other polypeptide.
[0299] In a proliferation assay, the STOP-1 polypeptide has the ability to stimulate the proliferation of endothelial cells in the presence of the co-mitogen ConA. Specifically, human umbilical vein endothelial cells can be obtained and cultured in 96-well flat-bottomed culture plates (Costar, Cambridge, Mass.) and supplemented with a reaction mixture appropriate for facilitating proliferation of the cells, the mixture containing Con-A (Calbiochem, La Jolla, Calif.). Con-A and the test inhibitory compound to be screened are added and after incubation at 37° C., cultures are pulsed with 3-H-thymidine and harvested onto glass fiber filters (phD; Cambridge Technology, Watertown, Mass.). Mean 3-H-thymidine incorporation (cpm) of triplicate cultures is determined using a liquid scintillation counter (Beckman Instruments, Irvine, Calif.). Significant 3-(H)-thymidine incorporation indicates stimulation of endothelial cell proliferation.
[0300] According to one embodiment, the assay described above or assays as described in the Examples below are performed to test antagonists of this invention. Alternatively, antagonists can be detected by combining the STOP-1 polypeptide and a potential antagonist with cold STOP-1 polypeptide under appropriate conditions for a competitive inhibition assay. The STOP-1 polypeptide can be labeled, such as by radioactivity or a colorimetric method, such that the number of STOP-1 polypeptide molecules bound can be used to determine the effectiveness of the potential antagonist. The STOP-1 polypeptide can be labeled by a variety of means including iodination or inclusion of a recognition site for a site-specific protein kinase. Following fixation and incubation, the slides are subjected to autoradiographic analysis
[0301] Drug candidates include anti-STOP-1 antibodies including, without limitation, poly- and monoclonal antibodies and antibody fragments, single-chain antibodies, anti-idiotypic antibodies, and chimeric or humanized versions of such antibodies or fragments, as well as human antibodies and antibody fragments. Alternatively, a drug candidate can be a closely related protein, for example, a mutated form of the STOP-1 polypeptide that competitively inhibits the action of the STOP-1 polypeptide.
Administration Protocols, Schedules, Doses, and Formulations
[0302] The molecules herein and antagonists thereto are pharmaceutically useful as a prophylactic and therapeutic agent for various disorders and diseases as set forth above.
[0303] Therapeutic compositions of the polypeptides, antibodies or antagonists of this invention are prepared for storage by mixing the desired molecule having the appropriate degree of purity with optional pharmaceutically acceptable carriers, excipients, or stabilizers (Remington's Pharmaceutical Sciences, 16th edition, Osol, A. ed. (1980)), in the form of lyophilized formulations or aqueous solutions. Acceptable carriers, excipients, or stabilizers are nontoxic to recipients at the dosages and concentrations employed, and include buffers such as phosphate, citrate, and other organic acids; antioxidants including ascorbic acid and methionine; preservatives (such as octadecyldimethylbenzyl ammonium chloride; hexamethonium chloride; benzalkonium chloride, benzethonium chloride; phenol, butyl or benzyl alcohol; alkyl parabens such as methyl or propyl paraben; catechol; resorcinol; cyclohexanol; 3-pentanol; and m-cresol); low molecular weight (less than about 10 residues) polypeptides; proteins, such as serum albumin, gelatin, or immunoglobulins; hydrophilic polymers such as polyvinylpyrrolidone; amino acids such as glycine, glutamine, asparagine, histidine, arginine, or lysine; monosaccharides, disaccharides, and other carbohydrates including glucose, mannose, or dextrins; chelating agents such as EDTA; sugars such as sucrose, mannitol, trehalose or sorbitol; salt-forming counter-ions such as sodium; metal complexes (e.g., Zn-protein complexes); and/or non-ionic surfactants such as TWEEN®, PLURONICS® or polyethylene glycol (PEG).
[0304] Additional examples of such carriers include ion exchangers, alumina, aluminum stearate, lecithin, serum proteins, such as human serum albumin, buffer substances such as phosphates, glycine, sorbic acid, potassium sorbate, partial glyceride mixtures of saturated vegetable fatty acids, water, salts, or electrolytes such as protamine sulfate, disodium hydrogen phosphate, potassium hydrogen phosphate, sodium chloride, zinc salts, colloidal silica, magnesium trisilicate, polyvinyl pyrrolidone, cellulose-based substances, and polyethylene glycol. Carriers for topical or gel-based forms of agonist or antagonist include polysaccharides such as sodium carboxymethylcellulose or methylcellulose, polyvinylpyrrolidone, polyacrylates, polyoxyethylene-polyoxypropylene-block polymers, polyethylene glycol, and wood wax alcohols. For all administrations, conventional depot forms are suitably used. Such forms include, for example, microcapsules, nano-capsules, liposomes, plasters, inhalation forms, nose sprays, sublingual tablets, and sustained-release preparations. The STOP-1 polypeptides or agonists or antagonists will typically be formulated in such vehicles at a concentration of about 0.1 mg/ml to 100 mg/ml.
[0305] Another formulation comprises incorporating a STOP-1 polypeptide or agonist or antagonist thereof into formed articles. Such articles can be used in modulating endothelial cell growth and angiogenesis. In addition, tumor invasion and metastasis can be modulated with these articles.
[0306] STOP-1 polypeptides or agonists or antagonists to be used for in vivo administration must be sterile. This is readily accomplished by filtration through sterile filtration membranes, prior to or following lyophilization and reconstitution. STOP-1 polypeptides ordinarily will be stored in lyophilized form or in solution if administered systemically. If in lyophilized form, the STOP-1 polypeptide or agonist or antagonist thereto is typically formulated in combination with other ingredients for reconstitution with an appropriate diluent at the time for use. An example of a liquid formulation of a STOP-1 polypeptide or agonist or antagonist is a sterile, clear, colorless unpreserved solution filled in a single-dose vial for subcutaneous injection. Preserved pharmaceutical compositions suitable for repeated use can contain, for example, depending mainly on the indication and type of polypeptide: [0307] a. STOP-1 polypeptide or agonist or antagonist thereto; [0308] b. a buffer capable of maintaining the pH in a range of maximum stability of the polypeptide or other molecule in solution, preferably about 4-8; [0309] c. a detergent/surfactant primarily to stabilize the polypeptide or molecule against agitation-induced aggregation; [0310] d. an isotonifier; [0311] e. a preservative selected from the group of phenol, benzyl alcohol and a benzethonium halide, e.g., chloride; and [0312] f. water.
[0313] If the detergent employed is non-ionic, it can, for example, be polysorbates (e.g., POLYSORBATE® (TWEEN®) 20, 80, etc.) or poloxamers (e.g., POLOXAMER® 188). The use of non-ionic surfactants permits the formulation to be exposed to shear surface stresses without causing denaturation of the polypeptide. Further, such surfactant-containing formulations can be employed in aerosol devices such as those used in a pulmonary dosing, and needleless jet injector guns (see, e.g., EP 257,956).
[0314] An isotonifier can be present to ensure isotonicity of a liquid composition of the STOP-1 polypeptide or agonist or antagonist thereto, and includes polyhydric sugar alcohols, preferably trihydric or higher sugar alcohols, such as glycerin, erythritol, arabitol, xylitol, sorbitol, and mannitol. These sugar alcohols can be used alone or in combination. Alternatively, sodium chloride or other appropriate inorganic salts can be used to render the solutions isotonic.
[0315] The buffer can, for example, be an acetate, citrate, succinate, or phosphate buffer depending on the pH desired. The pH of one type of liquid formulation of this invention is buffered in the range of about 4 to 8, preferably about physiological pH.
[0316] The preservatives phenol, benzyl alcohol and benzethonium halides, e.g., chloride, are known antimicrobial agents that can be employed.
[0317] Therapeutic STOP-1 polypeptide or antibody compositions generally are placed into a container having a sterile access port, for example, an intravenous solution bag or vial having a stopper pierceable by a hypodermic injection needle. The formulations are preferably administered as repeated intravenous (i.v.), subcutaneous (s.c.), or intramuscular (i.m.) injections, or as aerosol formulations suitable for intranasal or intrapulmonary delivery (for intrapulmonary delivery see, e.g., EP 257,956).
[0318] STOP-1 polypeptides or antibodies can also be administered in the form of sustained-released preparations. Suitable examples of sustained-release preparations include semipermeable matrices of solid hydrophobic polymers containing the protein, which matrices are in the form of shaped articles, e.g., films, or microcapsules. Examples of sustained-release matrices include polyesters, hydrogels (e.g., poly(2-hydroxyethyl-methacrylate) as described by Langer et al., J. Biomed. Mater. Res., 15: 167-277 (1981) and Langer, Chem. Tech., 12: 98-105 (1982) or poly(vinylalcohol)), polylactides (U.S. Pat. No. 3,773,919, EP 58,481), copolymers of L-glutamic acid and gamma ethyl-L-glutamate (Sidman et al., Biopolymers, 22: 547-556 (1983)), non-degradable ethylene-vinyl acetate (Langer et al., supra), degradable lactic acid-glycolic acid copolymers such as the Lupron Depot® (injectable microspheres composed of lactic acid-glycolic acid copolymer and leuprolide acetate), and poly-D-(-)-3-hydroxybutyric acid (EP 133,988).
[0319] While polymers such as ethylene-vinyl acetate and lactic acid-glycolic acid enable release of molecules for over 100 days, certain hydrogels release proteins for shorter time periods. When encapsulated proteins remain in the body for a long time, they can denature or aggregate as a result of exposure to moisture at 37° C., resulting in a loss of biological activity and possible changes in immunogenicity. Rational strategies can be devised for protein stabilization depending on the mechanism involved. For example, if the aggregation mechanism is discovered to be intermolecular S--S bond formation through thio-disulfide interchange, stabilization can be achieved by modifying sulfhydryl residues, lyophilizing from acidic solutions, controlling moisture content, using appropriate additives, and developing specific polymer matrix compositions.
[0320] Sustained-release STOP-1 polypeptide and antibody compositions also include liposomally entrapped STOP-1 polypeptides. Liposomes containing the STOP-1 polypeptide are prepared by methods known per se: DE 3,218,121; Epstein et al., Proc. Natl. Acad. Sci. USA, 82: 3688-3692 (1985); Hwang et al., Proc. Natl. Acad. Sci. USA, 77: 4030-4034 (1980); EP 52,322; EP 36,676; EP 88,046; EP 143,949; EP 142,641; Japanese patent application 83-118008; U.S. Pat. Nos. 4,485,045 and 4,544,545; and EP 102,324. Ordinarily the liposomes are of the small (about 200-800 Angstroms) unilamellar type in which the lipid content is greater than about 30 mol. % cholesterol, the selected proportion being adjusted for the optimal therapy.
[0321] The therapeutically effective dose of a STOP-1 polypeptide or antagonist thereto will, of course, vary depending on such factors as the proliferative disorder to be treated (including prevention), the method of administration, the type of compound being used for treatment, any co-therapy involved, the patient's age, weight, general medical condition, medical history, etc., and its determination is well within the skill of a practicing physician. Accordingly, it will be necessary for the therapist to titer the dosage and modify the route of administration as required to obtain the maximal therapeutic effect. The clinician will administer the STOP-1 polypeptide or antagonist until a dosage is reached that achieves the desired effect for treatment of the condition in question. For example, if the objective is the treatment of cancer, the amount would be one that inhibits the growth of the cancer.
[0322] With the above guidelines, the effective dose generally is within the range of from about 0.001 to about 1.0 mg/kg, more preferably about 0.01-1.0 mg/kg, most preferably about 0.01-0.1 mg/kg.
[0323] For non-oral use in treating proliferative disorders, it is advantageous to administer the STOP-1 polypeptide or antagonist thereto in the form of an injection at about 0.01 to 50 mg, preferably about 0.05 to 20 mg, most preferably 1 to 20 mg, per kg body weight, 1 to 3 times daily by intravenous injection. For oral administration, a molecule based on the STOP-1 polypeptide is preferably administered at about 5 mg to 1 g, preferably about 10 to 100 mg, per kg body weight, 1 to 3 times daily. It should be appreciated that endotoxin contamination should be kept minimally at a safe level, for example, less than 0.5 ng/mg protein. Moreover, for human administration, the formulations preferably meet sterility, pyrogenicity, general safety, and purity as required by FDA Office and Biologics standards.
[0324] The dosage regimen of a pharmaceutical composition containing the STOP-1 polypeptide to be used in tissue regeneration will be determined by the attending physician considering various factors that modify the action of the polypeptides, e.g., amount of tissue weight desired to be formed, the site of damage, the condition of the damaged tissue, the size of a wound, type of damaged tissue (e.g., bone), the patient's age, sex, and diet, the severity of any infection, time of administration, and other clinical factors. The dosage can vary with the type of matrix used in the reconstitution and with inclusion of other proteins in the pharmaceutical composition. For example, the addition of other known growth factors, such as IGF-I, to the final composition can also affect the dosage. Progress can be monitored by periodic assessment of tissue/bone growth and/or repair, for example, X-rays, histomorphometric determinations, and tetracycline labeling.
[0325] The route of STOP-1 polypeptide or antagonist or agonist administration is in accord with known methods, e.g., by injection or infusion by intravenous, intramuscular, intracerebral, intraperitoneal, intracerobrospinal, subcutaneous, intraocular, intraarticular, intrasynovial, intrathecal, oral, topical, or inhalation routes, or by sustained-release systems as noted below. The STOP-1 polypeptide or agonist or antagonists thereof also are suitably administered by intratumoral, peritumoral, intralesional, or perilesional routes, to exert local as well as systemic therapeutic effects. The intraperitoneal route is expected to be particularly useful, for example, in the treatment of ovarian tumors.
[0326] If a peptide or small molecule is employed as an antagonist or agonist, it is preferably administered orally or non-orally in the form of a liquid or solid to mammals.
[0327] Examples of pharmacologically acceptable salts of molecules that form salts and are useful hereunder include alkali metal salts (e.g., sodium salt, potassium salt), alkaline earth metal salts (e.g., calcium salt, magnesium salt), ammonium salts, organic base salts (e.g., pyridine salt, triethylamine salt), inorganic acid salts (e.g., hydrochloride, sulfate, nitrate), and salts of organic acid (e.g., acetate, oxalate, p-toluenesulfonate).
[0328] For compositions herein that are useful for bone, cartilage, tendon, or ligament regeneration, the therapeutic method includes administering the composition topically, systemically, or locally as an implant or device. When administered, the therapeutic composition for use is in a pyrogen-free, physiologically acceptable form. Further, the composition can desirably be encapsulated or injected in a viscous form for delivery to the site of bone, cartilage, or tissue damage. Topical administration can be suitable for wound healing and tissue repair. Preferably, for bone and/or cartilage formation, the composition would include a matrix capable of delivering the protein-containing composition to the site of bone and/or cartilage damage, providing a structure for the developing bone and cartilage and preferably capable of being resorbed into the body. Such matrices can be formed of materials presently in use for other implanted medical applications.
[0329] The choice of matrix material is based on biocompatibility, biodegradability, mechanical properties, cosmetic appearance, and interface properties. The particular application of the compositions will define the appropriate formulation. Potential matrices for the compositions can be biodegradable and chemically defined calcium sulfate, tricalcium phosphate, hydroxyapatite, polylactic acid, polyglycolic acid, and polyanhydrides. Other potential materials are biodegradable and biologically well-defined, such as bone or dermal collagen. Further matrices are comprised of pure proteins or extracellular matrix components. Other potential matrices are nonbiodegradable and chemically defined, such as sintered hydroxyapatite, bioglass, aluminates, or other ceramics. Matrices can be comprised of combinations of any of the above-mentioned types of material, such as polylactic acid and hydroxyapatite or collagen and tricalcium phosphate. The bioceramics can be altered in composition, such as in calcium-aluminate-phosphate and processing to alter pore size, particle size, particle shape, and biodegradability.
[0330] One specific embodiment is a 50:50 (mole weight) copolymer of lactic acid and glycolic acid in the form of porous particles having diameters ranging from 150 to 800 microns. In some applications, it will be useful to utilize a sequestering agent, such as carboxymethyl cellulose or autologous blood clot, to prevent the polypeptide compositions from disassociating from the matrix.
[0331] One suitable family of sequestering agents is cellulosic materials such as alkylcelluloses (including hydroxyalkylcelluloses), including methylcellulose, ethylcellulose, hydroxyethylcellulose, hydroxypropylcellulose, hydroxypropylmethylcellulose, and carboxymethylcellulose, one preferred being cationic salts of carboxymethylcellulose (CMC). Other preferred sequestering agents include hyaluronic acid, sodium alginate, poly(ethylene glycol), polyoxyethylene oxide, carboxyvinyl polymer, and poly(vinyl alcohol). The amount of sequestering agent useful herein is 0.5-20 wt %, preferably 1-10 wt %, based on total formulation weight, which represents the amount necessary to prevent desorption of the polypeptide (or its antagonist) from the polymer matrix and to provide appropriate handling of the composition, yet not so much that the progenitor cells are prevented from infiltrating the matrix, thereby providing the polypeptide (or its antagonist) the opportunity to assist the osteogenic activity of the progenitor cells.
Combination Therapies
[0332] The effectiveness of the STOP-1 polypeptide or an agonist or antagonist thereof in preventing or treating the disorder in question can be improved by administering the active agent serially or in combination with another agent that is effective for those purposes, either in the same composition or as separate compositions.
[0333] For example, for treatment of cell proliferative disorders, STOP-1 polypeptide antagonist therapy can be combined with the administration of other inhibitors of cell proliferation, such as cytotoxic agents.
[0334] In addition, STOP-1 polypeptide antagonists used to treat cancer can be combined with cytotoxic, chemotherapeutic, or growth-inhibitory agents as identified above. Also, for cancer treatment, the STOP-1 polypeptide antagonist thereof is suitably administered serially or in combination with radiological treatments, whether involving irradiation or administration of radioactive substances.
[0335] If the treating is for cancer, it may be desirable also to administer antibodies against other tumor-associated antigens, such as antibodies that bind to one or more of the ErbB2, EGFR, ErbB3, ErbB4, or VEGF receptor(s). Alternatively, or in addition, two or more antibodies binding the same or two or more different antigens disclosed herein may be co-administered to the patient. Sometimes, it may be beneficial also to administer one or more cytokines to the patient. In one preferred embodiment, the antagonist antibodies herein are co-administered with a growth-inhibitory agent. For example, the growth-inhibitory agent may be administered first, followed by an antagonist antibody of the present invention. However, simultaneous administration or administration of the antagonist antibody of the present invention first is also contemplated. Suitable dosages for the growth-inhibitory agent are those presently used and may be lowered due to the combined action (synergy) of the growth-inhibitory agent and the antibody herein.
[0336] In one embodiment, vascularization of tumors is attacked in combination therapy. The antagonist antibodies of this invention and another antibody (e.g., anti-VEGF) are administered to tumor-bearing patients at therapeutically effective doses as determined, for example, by observing necrosis of the tumor or its metastatic foci, if any. This therapy is continued until such time as no further beneficial effect is observed or clinical examination shows no trace of the tumor or any metastatic foci. Then TNF is administered, alone or in combination with an auxiliary agent such as alpha-, beta-, or gamma-interferon, anti-HER2 antibody, heregulin, anti-heregulin antibody, D-factor, interleukin-1 (IL-1), interleukin-2 (IL-2), granulocyte-macrophage colony stimulating factor (GM-CSF), or agents that promote microvascular coagulation in tumors, such as anti-protein C antibody, anti-protein S antibody, or C4b binding protein (see, WO 91/01753, published 21 Feb. 1991), or heat or radiation.
[0337] The effective amounts of the therapeutic agents administered in combination with the STOP-1 polypeptide or antagonist thereof will be at the physician's or veterinarian's discretion. Dosage administration and adjustment is done to achieve maximal management of the conditions to be treated. The dose will additionally depend on such factors as the type of the therapeutic agent to be used and the specific patient being treated. Typically, the amount employed will be the same dose as that used, if the given therapeutic agent is administered without the STOP-1 polypeptide.
Articles of Manufacture
[0338] An article of manufacture such as a kit containing the STOP-1 polypeptide or agonists or antagonists thereof useful for the diagnosis or treatment of the disorders described above comprises at least a container and a label. Suitable containers include, for example, bottles, vials, syringes, and test tubes. The containers can be formed from a variety of materials such as glass or plastic. The container holds a composition that is effective for diagnosing or treating the condition and can have a sterile access port (for example, the container can be an intravenous solution bag or a vial having a stopper pierceable by a hypodermic injection needle). The active agent in the composition is the STOP-1 polypeptide or an agonist or antagonist thereto. The label on, or associated with, the container indicates that the composition is used for diagnosing or treating the condition of choice. The article of manufacture can further comprise a second container comprising a pharmaceutically-acceptable buffer, such as phosphate-buffered saline, Ringer's solution, and dextrose solution. It can further include other materials desirable from a commercial and user standpoint, including other buffers, diluents, filters, needles, syringes, and package inserts with instructions for use. The article of manufacture can also comprise a second or third container with another active agent as described above.
Polyclonal Antibodies
[0339] Methods of preparing polyclonal antibodies are known to the skilled artisan. Polyclonal antibodies can be raised in a mammal, for example, by one or more injections of an immunizing agent and, if desired, an adjuvant. Typically, the immunizing agent and/or adjuvant will be injected in the mammal by multiple subcutaneous or intraperitoneal injections. The immunizing agent can include the STOP-1 polypeptide or a fusion protein thereof. It can be useful to conjugate the immunizing agent to a protein known to be immunogenic in the mammal being immunized. Examples of such immunogenic proteins include, but are not limited to, keyhole limpet hemocyanin, serum albumin, bovine thyroglobulin, and soybean trypsin inhibitor. Examples of adjuvants that can be employed include Freund's complete adjuvant and MPL-TDM adjuvant (monophosphoryl Lipid A or synthetic trehalose dicorynomycolate). The immunization protocol can be selected by one skilled in the art without undue experimentation.
Monoclonal Antibodies
[0340] Anti-STOP-1 antibodies can be monoclonal antibodies. Monoclonal antibodies can be prepared, e.g., using hybridoma methods, such as those described by Kohler and Milstein, Nature, 256:495 (1975) or can be made by recombinant DNA methods (U.S. Pat. No. 4,816,567) or can be produced by the methods described herein in the Example section. In a hybridoma method, a mouse, hamster, or other appropriate host animal is typically immunized with an immunizing agent to elicit lymphocytes that produce or are capable of producing antibodies that will specifically bind to the immunizing agent. Alternatively, the lymphocytes can be immunized in vitro.
[0341] The immunizing agent will typically include the STOP-1 polypeptide or a fusion protein thereof. Generally, either peripheral blood lymphocytes ("PBLs") are used if cells of human origin are desired, or spleen cells or lymph node cells are used if non-human mammalian sources are desired. The lymphocytes are then fused with an immortalized cell line using a suitable fusing agent, such as polyethylene glycol, to form a hybridoma cell. Goding, Monoclonal Antibodies: Principles and Practice (New York: Academic Press, 1986), pp. 59-103 Immortalized cell lines are usually transformed mammalian cells, particularly myeloma cells of rodent, bovine, and human origin. Usually, rat or mouse myeloma cell lines are employed. The hybridoma cells can be cultured in a suitable culture medium that preferably contains one or more substances that inhibit the growth or survival of the unfused, immortalized cells. For example, if the parental cells lack the enzyme hypoxanthine guanine phosphoribosyl transferase (HGPRT or HPRT), the culture medium for the hybridomas typically will include hypoxanthine, aminopterin, and thymidine ("HAT medium"), which substances prevent the growth of HGPRT-deficient cells.
[0342] Preferred immortalized cell lines are those that fuse efficiently, support stable high-level expression of antibody by the selected antibody-producing cells, and are sensitive to a medium such as HAT medium. More preferred immortalized cell lines are murine myeloma lines, which can be obtained, for instance, from the Salk Institute Cell Distribution Center, San Diego, Calif. and the American Type Culture Collection, Manassas, Va. Human myeloma and mouse-human heteromyeloma cell lines also have been described for the production of human monoclonal antibodies. Kozbor, J. Immunol., 133:3001 (1984); Brodeur et al., Monoclonal Antibody Production Techniques and Applications (Marcel Dekker, Inc.: New York, 1987) pp. 51-63.
[0343] The culture medium in which the hybridoma cells are cultured can then be assayed for the presence of monoclonal antibodies directed against the STOP-1 polypeptide. Preferably, the binding specificity of monoclonal antibodies produced by the hybridoma cells is determined by immunoprecipitation or by an in vitro binding assay, such as radioimmunoassay (RIA) or enzyme-linked immunoabsorbent assay (ELISA). Such techniques and assays are known in the art. The binding affinity of the monoclonal antibody can, for example, be determined by the Scatchard analysis of Munson and Pollard, Anal. Biochem., 107:220 (1980).
[0344] After the desired hybridoma cells are identified, the clones can be subcloned by limiting dilution procedures and grown by standard methods. Goding, supra. Suitable culture media for this purpose include, for example, Dulbecco's Modified Eagle's Medium and RPMI-1640 medium. Alternatively, the hybridoma cells can be grown in vivo as ascites in a mammal.
[0345] The monoclonal antibodies secreted by the subclones can be isolated or purified from the culture medium or ascites fluid by conventional immunoglobulin purification procedures such as, for example, protein A-Sepharose, hydroxylapatite chromatography, gel electrophoresis, dialysis, or affinity chromatography.
[0346] The monoclonal antibodies can also be made by recombinant DNA methods, such as those described in U.S. Pat. No. 4,816,567. DNA encoding the monoclonal antibodies of the invention can be readily isolated and sequenced using conventional procedures (e.g., by using oligonucleotide probes that are capable of binding specifically to genes encoding the heavy and light chains of murine antibodies). The hybridoma cells of the invention serve as a preferred source of such DNA. Once isolated, the DNA can be placed into expression vectors, which are then transfected into host cells such as simian COS cells, Chinese hamster ovary (CHO) cells, or myeloma cells that do not otherwise produce immunoglobulin protein, to obtain the synthesis of monoclonal antibodies in the recombinant host cells. The DNA also can be modified, for example, by substituting the coding sequence for human heavy- and light-chain constant domains in place of the homologous murine sequences (U.S. Pat. No. 4,816,567; Morrison et al., supra) or by covalently joining to the immunoglobulin coding sequence all or part of the coding sequence for a non-immunoglobulin polypeptide. Such a non-immunoglobulin polypeptide can be substituted for the constant domains of an antibody of the invention, or can be substituted for the variable domains of one antigen-combining site of an antibody of the invention to create a chimeric bivalent antibody.
[0347] The antibodies can be monovalent antibodies. Methods for preparing monovalent antibodies are known in the art. For example, one method involves recombinant expression of immunoglobulin light chain and modified heavy chain. The heavy chain is truncated generally at any point in the Fc region so as to prevent heavy-chain crosslinking. Alternatively, the relevant cysteine residues are substituted with another amino acid residue or are deleted so as to prevent crosslinking.
[0348] In vitro methods are also suitable for preparing monovalent antibodies. Digestion of antibodies to produce fragments thereof, particularly Fab fragments, can be accomplished using techniques known in the art.
Human and Humanized Antibodies
[0349] The anti-STOP-1 antibodies can further comprise humanized antibodies or human antibodies. Humanized forms of non-human (e.g., murine) antibodies are chimeric immunoglobulins, immunoglobulin chains, or fragments thereof (such as Fv, Fab, Fab', F(ab')2, or other antigen-binding subsequences of antibodies) that contain minimal sequence derived from non-human immunoglobulin. Humanized antibodies include human immunoglobulins (recipient antibody) in which residues from a CDR of the recipient are replaced by residues from a CDR of a non-human species (donor antibody) such as mouse, rat, or rabbit having the desired specificity, affinity, and capacity. In some instances, Fv framework residues of the human immunoglobulin are replaced by corresponding non-human residues. Humanized antibodies can also comprise residues that are found neither in the recipient antibody nor in the imported CDR or framework sequences. In general, the humanized antibody will comprise substantially all of at least one, and typically two, variable domains, in which all or substantially all of the CDR regions correspond to those of a non-human immunoglobulin, and all or substantially all of the FR regions are those of a human immunoglobulin consensus sequence. The humanized antibody preferably also will comprise at least a portion of an immunoglobulin constant region (Fc), typically that of a human immunoglobulin. Jones et al., Nature, 321: 522-525 (1986); Riechmann et al., Nature, 332: 323-329 (1988); Presta, Curr. Op. Struct. Biol., 2:593-596 (1992).
[0350] Methods for humanizing non-human antibodies are known in the art. Generally, a humanized antibody has one or more amino acid residues introduced into it from a source that is non-human. These non-human amino acid residues are often referred to as "import" residues, which are typically taken from an "import" variable domain. Humanization can be essentially performed following the method of Winter and co-workers (Jones et al., Nature, 321: 522-525 (1986); Riechmann et al., Nature, 332: 323-327 (1988); Verhoeyen et al., Science, 239: 1534-1536 (1988)), by substituting rodent CDRs or CDR sequences for the corresponding sequences of a human antibody. Accordingly, such "humanized" antibodies are chimeric antibodies (U.S. Pat. No. 4,816,567), wherein substantially less than an intact human variable domain has been substituted by the corresponding sequence from a non-human species. In practice, humanized antibodies are typically human antibodies in which some CDR residues and possibly some FR residues are substituted by residues from analogous sites in rodent antibodies.
[0351] As an alternative to humanization, human antibodies can be generated. For example, it is now possible to produce transgenic animals (e.g., mice) that are capable, upon immunization, of producing a full repertoire of human antibodies in the absence of endogenous immunoglobulin production. For example, it has been described that the homozygous deletion of the antibody heavy-chain joining region (JH) gene in chimeric and germ-line mutant mice results in complete inhibition of endogenous antibody production. Transfer of the human germ-line immunoglobulin gene array into such germ-line mutant mice will result in the production of human antibodies upon antigen challenge. See, e.g., Jakobovits et al., Proc. Natl. Acad. Sci. USA, 90:2551 (1993); Jakobovits et al., Nature, 362:255-258 (1993); Bruggemann et al., Year in Immuno, 7:33 (1993); U.S. Pat. Nos. 5,545,806, 5,569,825, 5,591,669 (all of GenPharm); 5,545,807; and WO 97/17852. Alternatively, human antibodies can be made by introducing human immunoglobulin loci into transgenic animals, e.g., mice in which the endogenous immunoglobulin genes have been partially or completely inactivated. Upon challenge, human antibody production is observed that closely resembles that seen in humans in all respects, including gene rearrangement, assembly, and antibody repertoire. This approach is described, for example, in U.S. Pat. Nos. 5,545,807; 5,545,806; 5,569,825; 5,625,126; 5,633,425; and 5,661,016, and in the following scientific publications: Marks et al., Bio/Technology, 10: 779-783 (1992); Lonberg et al., Nature, 368: 856-859 (1994); Morrison, Nature, 368: 812-813 (1994); Fishwild et al., Nature Biotechnology, 14: 845-851 (1996); Neuberger, Nature Biotechnology, 14: 826 (1996); Lonberg and Huszar, Intern. Rev. Immunol., 13: 65-93 (1995).
[0352] Alternatively, phage display technology (McCafferty et al., Nature 348:552-553 [1990]) can be used to produce human antibodies and antibody fragments in vitro, from immunoglobulin variable (V) domain gene repertoires from unimmunized donors. According to this technique, antibody V domain genes are cloned in-frame into either a major or minor coat protein gene of a filamentous bacteriophage, such as M13 or fd, and displayed as functional antibody fragments on the surface of the phage particle. Because the filamentous particle contains a single-stranded DNA copy of the phage genome, selections based on the functional properties of the antibody also result in selection of the gene encoding the antibody exhibiting those properties. Thus, the phage mimics some of the properties of the B-cell. Phage display can be performed in a variety of formats, reviewed in, e.g., Johnson, Kevin S, and Chiswell, David J., Current Opinion in Structural Biology 3:564-571 (1993). Several sources of V-gene segments can be used for phage display. Clackson et al., Nature, 352:624-628 (1991) isolated a diverse array of anti-oxazolone antibodies from a small random combinatorial library of V genes derived from the spleens of immunized mice. A repertoire of V genes from unimmunized human donors can be constructed and antibodies to a diverse array of antigens (including self-antigens) can be isolated essentially following the techniques described by Marks et al., J. Mol. Biol. 222:581-597 (1991), or Griffith et al., EMBO J. 12:725-734 (1993). See, also, U.S. Pat. Nos. 5,565,332 and 5,573,905.
[0353] As discussed above, human antibodies may also be generated by in vitro activated B cells (see U.S. Pat. Nos. 5,567,610 and 5,229,275).
[0354] Human antibodies can also be produced using various techniques known in the art, including phage display libraries. Hoogenboom and Winter, J. Mol. Biol., 227: 381 (1991); Marks et al., J. Mol. Biol., 222: 581 (1991). The techniques of Cole et al. and Boerner et al. are also available for the preparation of human monoclonal antibodies. Cole et al., Monoclonal Antibodies and Cancer Therapy, Alan R. Liss, p. 77 (1985) and Boerner et al., J. Immunol., 147(1): 86-95 (1991).
Bispecific Anti-STOP-1 Antibodies
[0355] Bispecific antibodies are monoclonal, preferably human or humanized, antibodies that have binding specificities for at least two different antigens. In the present case, one of the binding specificities is for the STOP-1 polypeptide, the other one is for any other antigen, and preferably for a cell-surface protein or receptor or receptor subunit. For example, the cell-surface protein can be a natural killer (NK) cell receptor. Thus, according to one embodiment, a bispecific antibody of this invention can bind STOP-1 and bind a NK cell and, optionally, activate the NK cell. According to another embodiment, a bispecific antibody of this invention can bind STOP-1 and binds to a stromal tissue compared to other tissue (e.g., stromal targeting agent).
[0356] Methods for making bispecific antibodies are known in the art. Traditionally, the recombinant production of bispecific antibodies is based on the co-expression of two immunoglobulin heavy-chain/light-chain pairs, where the two heavy chains have different specificities. Milstein and Cuello, Nature, 305: 537-539 (1983). Because of the random assortment of immunoglobulin heavy and light chains, these hybridomas (quadromas) produce a potential mixture of ten different antibody molecules, of which only one has the correct bispecific structure. The purification of the correct molecule is usually accomplished by affinity chromatography steps. Similar procedures are disclosed in WO 93/08829, published 13 May 1993, and in Traunecker et al., EMBO J., 10: 3655-3659 (1991).
[0357] Antibody variable domains with the desired binding specificities (antibody-antigen combining sites) can be fused to immunoglobulin constant-domain sequences. The fusion preferably is with an immunoglobulin heavy-chain constant domain, comprising at least part of the hinge, CH2, and CH3 regions. It is preferred to have the first heavy-chain constant region (CH1) containing the site necessary for light-chain binding present in at least one of the fusions. DNAs encoding the immunoglobulin heavy-chain fusions and, if desired, the immunoglobulin light chain, are inserted into separate expression vectors, and are co-transfected into a suitable host organism. For further details of generating bispecific antibodies, see, for example, Suresh et al., Methods in Enzymology, 121: 210 (1986).
[0358] Various techniques for making and isolating bispecific antibody fragments directly from recombinant cell culture have also been described. For example, bispecific antibodies have been produced using leucine zippers. Kostelny et al., J. Immunol., 148(5):1547-1553 (1992). The leucine zipper peptides from the Fos and Jun proteins were linked to the Fab' portions of two different antibodies by gene fusion. The antibody homodimers were reduced at the hinge region to form monomers and then re-oxidized to form the antibody heterodimers. This method can also be utilized for the production of antibody homodimers. The "diabody" technology described by Hollinger et al., Proc. Natl. Acad. Sci. USA, 90:6444-6448 (1993) has provided an alternative mechanism for making bispecific antibody fragments. The fragments comprise a VH connected to a VL by a linker which is too short to allow pairing between the two domains on the same chain. Accordingly, the VH and VL domains of one fragment are forced to pair with the complementary VL and VH domains of another fragment, thereby forming two antigen-binding sites. Another strategy for making bispecific antibody fragments by the use of single-chain FY (sFv) dimers has also been reported. See Gruber et al., J. Immunol., 152:5368 (1994).
[0359] Antibodies with more than two valencies are contemplated. For example, trispecific antibodies can be prepared. Tutt et al. J. Immunol. 147: 60 (1991)
Heteroconjugate Antibodies
[0360] Heteroconjugate antibodies are composed of two covalently joined antibodies. Such antibodies have, for example, been proposed to target immune-system cells to unwanted cells (U.S. Pat. No. 4,676,980), and for treatment of HIV infection. WO 91/00360; WO 92/200373; EP 03089. It is contemplated that the antibodies can be prepared in vitro using known methods in synthetic protein chemistry, including those involving crosslinking agents. For example, immunotoxins can be constructed using a disulfide-exchange reaction or by forming a thioether bond. Examples of suitable reagents for this purpose include iminothiolate and methyl-4-mercaptobutyrimidate and those disclosed, for example, in U.S. Pat. No. 4,676,980.
Effector Function Engineering
[0361] It can be desirable to modify the antibody of the invention with respect to effector function, so as to enhance, e.g., the effectiveness of the antibody in treating cancer. For example, cysteine residue(s) can be introduced into the Fc region, thereby allowing interchain disulfide bond formation in this region. The homodimeric antibody thus generated can have improved internalization capability and/or increased complement-mediated cell killing and antibody-dependent cellular cytotoxicity (ADCC). See, Caron et al., J. Exp. Med., 176: 1191-1195 (1992) and Shopes, J. Immunol., 148: 2918-2922 (1992). Homodimeric antibodies with enhanced anti-tumor activity can also be prepared using heterobifunctional cross-linkers as described in Wolff et al., Cancer Research, 53: 2560-2565 (1993). Alternatively, an antibody can be engineered that has dual Fc regions and can thereby have enhanced complement lysis and ADCC capabilities. See, Stevenson et al., Anti-Cancer Drug Design, 3: 219-230 (1989).
Immunoconjugates
[0362] The invention also pertains to immunoconjugates comprising an antibody conjugated to a cytotoxic agent such as a chemotherapeutic agent, toxin (e.g., an enzymatically active toxin of bacterial, fungal, plant, or animal origin, or fragments thereof), or a radioactive isotope (i.e., a radioconjugate).
[0363] Chemotherapeutic agents useful in the generation of such immunoconjugates have been described above. Enzymatically active toxins and fragments thereof that can be used include diphtheria A chain, nonbinding active fragments of diphtheria toxin, exotoxin A chain (from Pseudomonas aeruginosa), ricin A chain, abrin A chain, modeccin A chain, alpha-sarcin, Aleurites fordii proteins, dianthin proteins, Phytolaca americana proteins (PAPI, PAPII, and PAP-S), momordica charantia inhibitor, curcin, crotin, sapaonaria officinalis inhibitor, gelonin, mitogellin, restrictocin, phenomycin, enomycin, and the tricothecenes. A variety of radionuclides are available for the production of radioconjugated antibodies. Examples include 212Bi, 131I, 131In, 90Y, and 186Re.
[0364] Conjugates of the antibody and cytotoxic agent are made using a variety of bifunctional protein-coupling agents such as N-succinimidyl-3-(2-pyridyldithiol) propionate (SPDP), iminothiolane (IT), bifunctional derivatives of imidoesters (such as dimethyl adipimidate HCl), active esters (such as disuccinimidyl suberate), aldehydes (such as glutaraldehyde), bis-azido compounds (such as bis (p-azidobenzoyl)hexanediamine), bis-diazonium derivatives (such as bis-(p-diazoniumbenzoyl)-ethylenediamine), diisocyanates (such as tolyene 2,6-diisocyanate), and bis-active fluorine compounds (such as 1,5-difluoro-2,4-dinitrobenzene). For example, a ricin immunotoxin can be prepared as described in Vitetta et al., Science, 238: 1098 (1987). Carbon-14-labeled 1-isothiocyanatobenzyl-3-methyldiethylene triaminepentaacetic acid (MX-DTPA) is an exemplary chelating agent for conjugation of radionucleotide to the antibody. See, WO94/11026.
[0365] In another embodiment, the antibody can be conjugated to a "receptor" (such as streptavidin) for utilization in tumor pretargeting wherein the antibody-receptor conjugate is administered to the patient, followed by removal of unbound conjugate from the circulation using a clearing agent and then administration of a "ligand" (e.g., avidin) that is conjugated to a cytotoxic agent (e.g., a radionucleotide).
Immunoliposomes
[0366] The antibodies disclosed herein can also be formulated as immunoliposomes. Liposomes containing the antibody are prepared by methods known in the art, such as described in Epstein et al., Proc. Natl. Acad. Sci. USA, 82: 3688 (1985); Hwang et al., Proc. Natl. Acad. Sci. USA, 77: 4030 (1980); and U.S. Pat. Nos. 4,485,045 and 4,544,545. Liposomes with enhanced circulation time are disclosed in U.S. Pat. No. 5,013,556.
[0367] Particularly useful liposomes can be generated by the reverse-phase evaporation method with a lipid composition comprising phosphatidylcholine, cholesterol, and PEG-derivatized phosphatidylethanolamine (PEG-PE). Liposomes are extruded through filters of defined pore size to yield liposomes with the desired diameter. Fab' fragments of the antibody of the present invention can be conjugated to the liposomes as described in Martin et al., J. Biol. Chem., 257: 286-288 (1982) via a disulfide-interchange reaction. A chemotherapeutic agent (such as Doxorubicin) is optionally contained within the liposome. See, Gabizon et al., J. National Cancer Inst., 81(19): 1484 (1989).
Pharmaceutical Compositions of Antibodies
[0368] Antibodies specifically binding a STOP-1 polypeptide identified herein, as well as other molecules identified by the screening assays disclosed hereinbefore, can be administered for the treatment of various disorders as noted above and below in the form of pharmaceutical compositions.
[0369] Lipofectins or liposomes can be used to deliver the polypeptides, nucleic acid molecules, antibodies, antagonists or composition of this invention into cells. Where antibody fragments are used, the smallest inhibitory fragment that specifically binds to the binding domain of the target protein is preferred. For example, based upon the variable-region sequences of an antibody, peptide molecules can be designed that retain the ability to bind the target protein sequence. Such peptides can be synthesized chemically and/or produced by recombinant DNA technology. See, e.g., Marasco et al., Proc. Natl. Acad. Sci. USA, 90: 7889-7893 (1993).
[0370] The formulation herein can also contain more than one active compound as necessary for the particular indication being treated, preferably those with complementary activities that do not adversely affect each other. Alternatively, or in addition, the composition can comprise an agent that enhances its function, such as, for example, a cytotoxic agent, chemotherapeutic agent, or growth-inhibitory agent. Such molecules are suitably present in combination in amounts that are effective for the purpose intended.
[0371] The active ingredients can also be entrapped in microcapsules prepared, for example, by coacervation techniques or by interfacial polymerization, for example, hydroxymethylcellulose or gelatin-microcapsules and poly-(methylmethacylate) microcapsules, respectively, in colloidal drug delivery systems (for example, liposomes, albumin microspheres, microemulsions, nano-particles, and nanocapsules) or in macroemulsions. Such techniques are disclosed in Remington's Pharmaceutical Sciences, supra.
[0372] The formulations to be used for in vivo administration must be sterile. This is readily accomplished by filtration through sterile filtration membranes.
[0373] Sustained-release preparations can be prepared. Suitable examples of sustained-release preparations include semipermeable matrices of solid hydrophobic polymers containing the antibody, which matrices are in the form of shaped articles, e.g., films, or microcapsules. Examples of sustained-release matrices include polyesters, hydrogels (for example, poly(2-hydroxyethyl-methacrylate), or poly(vinylalcohol)), polylactides (U.S. Pat. No. 3,773,919), copolymers of L-glutamic acid and ethyl-L-glutamate, non-degradable ethylene-vinyl acetate, degradable lactic acid-glycolic acid copolymers such as the LUPRON DEPOT® (injectable microspheres composed of lactic acid-glycolic acid copolymer and leuprolide acetate), and poly-D-(-)-3-hydroxybutyric acid. While polymers such as ethylene-vinyl acetate and lactic acid-glycolic acid enable release of molecules for over 100 days, certain hydrogels release proteins for shorter time periods. When encapsulated antibodies remain in the body for a long time, they can denature or aggregate as a result of exposure to moisture at 37° C., resulting in a loss of biological activity and possible changes in immunogenicity. Rational strategies can be devised for stabilization depending on the mechanism involved. For example, if the aggregation mechanism is discovered to be intermolecular S--S bond formation through thio-disulfide interchange, stabilization can be achieved by modifying sulfhydryl residues, lyophilizing from acidic solutions, controlling moisture content, using appropriate additives, and developing specific polymer matrix compositions.
Methods of Treatment using the anti-STOP-1 Antibody
[0374] It is contemplated that the antibodies to a STOP-1 polypeptide can be used to treat various proliferative disorders and diseases complicated or related to angiogenesis as noted above.
[0375] The antibodies are administered to a mammal, preferably a human, in accord with known methods, such as intravenous administration as a bolus or by continuous infusion over a period of time, by intramuscular, intraperitoneal, intracerobrospinal, subcutaneous, intra-articular, intrasynovial, intrathecal, oral, topical, or inhalation routes. Intravenous administration of the antibody is preferred.
[0376] Other therapeutic regimens can be combined with the administration of the antibodies of the instant invention as noted above. For example, if the antibodies are to treat cancer, the patient to be treated with such antibodies can also receive radiation therapy. Alternatively, or in addition, a chemotherapeutic agent can be administered to the patient. Preparation and dosing schedules for such chemotherapeutic agents can be used according to manufacturers' instructions or as determined empirically by the skilled practitioner. Preparation and dosing schedules for such chemotherapy are also described in Chemotherapy Service, Ed., M. C. Perry (Williams & Wilkins: Baltimore, Md., 1992). The chemotherapeutic agent can precede, or follow administration of the antibody, or can be given simultaneously therewith. The antibody can be combined with an anti-estrogen compound such as tamoxifen or EVISTA® or an anti-progesterone such as onapristone (see, EP 616812) in dosages known for such molecules.
[0377] If the antibodies are used for treating cancer, they can be, optionally, administer with antibodies against other tumor-associated antigens, such as antibodies that bind to one or more of the ErbB2, EGFR, ErbB3, ErbB4, or VEGF receptor(s). These also include the agents set forth above. Also, the antibody is suitably administered serially or in combination with radiological treatments, whether involving irradiation or administration of radioactive substances. Alternatively, or in addition, two or more antibodies binding the same or two or more different antigens disclosed herein can be co-administered to the patient. In a preferred embodiment, the antibodies herein are co-administered with a growth-inhibitory agent. For example, the growth-inhibitory agent can be administered first, followed by an antibody of the present invention. However, simultaneous administration or administration of the antibody of the present invention first is also contemplated. Suitable dosages for the growth-inhibitory agent are those presently used and can be lowered due to the combined action (synergy) of the growth-inhibitory agent and the antibody herein.
[0378] In one embodiment, vascularization of tumors is attacked in combination therapy. The anti-STOP-1 polypeptide antibody and another antibody (e.g., anti-VEGF) are administered to tumor-bearing patients at therapeutically effective doses as determined, for example, by observing necrosis of the tumor or its metastatic foci, if any. This therapy is continued until such time as no further beneficial effect is observed or clinical examination shows no trace of the tumor or any metastatic foci. Then TNF is administered, alone or in combination with an auxiliary agent such as alpha-, beta-, or gamma-interferon, anti-HER2 antibody, heregulin, anti-heregulin antibody, D-factor, interleukin-1 (IL-1), interleukin-2 (IL-2), granulocyte-macrophage colony stimulating factor (GM-CSF), or agents that promote microvascular coagulation in tumors, (such as anti-protein C antibody, anti-protein S antibody, or C4b binding protein, see, WO 91/01753, published 21 Feb. 1991), or heat or radiation.
[0379] Since the auxiliary agents will vary in their effectiveness, it can be desirable to compare their impact on the tumor by matrix screening in conventional fashion. The administration of anti-STOP-1 polypeptide antibody and TNF is repeated until the desired clinical effect is achieved. Alternatively, the anti-STOP-1 polypeptide antibody is administered together with TNF and, optionally, auxiliary agent(s). In instances where solid tumors are found in the limbs or in other locations susceptible to isolation from the general circulation, the therapeutic agents described herein are administered to the isolated tumor or organ. In other embodiments, a FGF or PDGF antagonist, such as an anti-FGF or an anti-PDGF neutralizing antibody, is administered to the patient in conjunction with the anti-STOP-1 polypeptide antibody. Treatment with anti-STOP-1 polypeptide antibodies preferably can be suspended during periods of wound healing or desirable neovascularization.
[0380] For the prevention or treatment of a proliferative disorder, the appropriate dosage of an antibody herein will depend on the type of disorder to be treated, as defined above, the severity and course of the disease, whether the antibody is administered for preventive or therapeutic purposes, previous therapy, the patient's clinical history and response to the antibody, and the discretion of the attending physician. The antibody is suitably administered to the patient at one time or over a series of treatments.
[0381] For example, depending on the type and severity of the disorder, about 1 ug/kg to 50 mg/kg (e.g., 0.1-20 mg/kg) of antibody is an initial candidate dosage for administration to the patient, whether, for example, by one or more separate administrations, or by continuous infusion. A typical daily or weekly dosage might range from about 1 ug/kg to 100 mg/kg or more, depending on the factors mentioned above. For repeated administrations over several days or longer, depending on the condition, the treatment is repeated or sustained until a desired suppression of disorder symptoms occurs. However, other dosage regimens can be useful. The progress of this therapy is easily monitored by conventional techniques and assays, including, for example, radiographic tumor imaging.
Articles of Manufacture with Antibodies
[0382] An article of manufacture containing a container with the antibody and a label is also provided. Such articles are described above, wherein the active agent is an anti-STOP-1 antibody.
Diagnosis and Prognosis of Tumors using Antibodies
[0383] If the indication for which the antibodies are used is cancer, while cell-surface proteins, such as growth receptors over expressed in certain tumors, are excellent targets for drug candidates or tumor (e.g., cancer) treatment, the same proteins along with STOP-1 polypeptides find additional use in the diagnosis and prognosis of tumors. For example, antibodies directed against the STOP-1 polypeptides can be used as tumor diagnostics or prognostics.
[0384] For example, antibodies, including antibody fragments, can be used qualitatively or quantitatively to detect the expression of genes including the gene encoding the STOP-1 polypeptide. The antibody preferably is equipped with a detectable, e.g., fluorescent label, and binding can be monitored by light microscopy, flow cytometry, fluorimetry, or other techniques known in the art. Such binding assays are performed essentially as described above.
[0385] In situ detection of antibody binding to the marker gene products can be performed, for example, by immunofluorescence or immunoelectron microscopy. For this purpose, a histological specimen is removed from the patient, and a labeled antibody is applied to it, preferably by overlaying the antibody on a biological sample. This procedure also allows for determining the distribution of the marker gene product in the tissue examined. It will be apparent to those skilled in the art that a wide variety of histological methods are readily available for in situ detection.
[0386] All publications (including patents and patent applications) cited herein are hereby incorporated in their entirety by reference.
[0387] The deposits herein were made under the provisions of the Budapest Treaty on the International Recognition of the Deposit of Microorganisms for the Purpose of Patent Procedure and the Regulations thereunder (Budapest Treaty). This assures maintenance of a viable culture of the deposits for 30 years from the date of deposit. The deposits will be made available by ATCC under the terms of the Budapest Treaty, and subject to an agreement between Genentech, Inc. and ATCC, which assures permanent and unrestricted availability of the progeny of the culture of the deposits to the public upon issuance of the pertinent U.S. patent or upon laying open to the public of any U.S. or foreign patent application, whichever comes first, and assures availability of the progeny to one determined by the U.S. Commissioner of Patents and Trademarks to be entitled thereto according to 35 U.S.C. 122 and the Commissioner's rules pursuant to thereto (including 37 C.F.R. 1.14 with particular reference to 886 OG 638).
[0388] The assignee of the present application has agreed that if a culture of the materials on deposits should die or be lost or destroyed when cultivated under suitable conditions, the materials will be promptly replaced on notification with another of the same. Availability of the deposited material is not to be construed as a license to practice the invention in contravention of the rights granted under the authority of any government in accordance with its patent laws.
[0389] Commercially available reagents referred to in the Examples were used according to manufacturer's instructions unless otherwise indicated. The source of those cells identified in the following Examples, and throughout the specification, by ATCC accession numbers is the American Type Culture Collection, Manassas, Va. Unless otherwise noted, the present invention uses standard procedures of recombinant DNA technology, such as those described hereinabove and in the following textbooks: Sambrook et al., supra; Ausubel et al., Current Protocols in Molecular Biology (Green Publishing Associates and Wiley Interscience, N.Y., 1989); Innis et al., PCR Protocols: A Guide to Methods and Applications (Academic Press, Inc.: N.Y., 1990); Harlow et al., Antibodies: A Laboratory Manual (Cold Spring Harbor Press: Cold Spring Harbor, 1988); Gait, Oligonucleotide Synthesis (IRL Press: Oxford, 1984); Freshney, Animal Cell Culture, 1987; Coligan et al., Current Protocols in Immunology, 1991.
[0390] Throughout this specification and claims, the word "comprise," or variations such as "comprises" or "comprising," will be understood to imply the inclusion of a stated integer or group of integers but not the exclusion of any other integer or group of integers.
[0391] The foregoing written description is considered to be sufficient to enable one skilled in the art to practice the invention. The following Examples are offered for illustrative purposes only, and are not intended to limit the scope of the present invention in any way. Indeed, various modifications of the invention in addition to those shown and described herein will become apparent to those skilled in the art from the foregoing description and fall within the scope of the appended claims.
EXPERIMENTAL SECTION
Example 1
STOP-1 is Conserved in Evolution
[0392] Nucleic acid molecules containing human, mouse and zebra fish STOP-1 were obtained by using PCR. Sequences with homology to human, mouse and zebra fish STOP-1 can be found in the Genebank database mouse EST: AK003674; chicken ESTs: A1585129, AL585130; rice fish ESTs: BJ490431, BJ498080, BJ510203, BJ504730; and zebra fish ESTs: AL727874, AW595388; and HGT AL844521 Amino acid sequences of human, mouse, rice fish, zebra fish and chicken STOP-1 nucleic acid molecules are described in SEQ ID NOS: 3, 4, 5, 6, and 7, respectively, and FIG. 1. The cDNAs of human STOP-1 were deposited under the terms of the Budapest Treaty with the American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, Va. 20110-2209, USA as described below:
TABLE-US-00013 Species Material Deposit No. Deposit Date Human 76393-1664 203323 Oct. 6, 1998 Human h762HIP PTA-5019 Feb. 21, 2003
[0393] Amino acid alignment of STOP-1 among several higher vertebrate species (human, mouse, chicken, zebra and rice fishes) shows a high degree of conservation, in particular, parts of the C-terminal domain including the triple helix domain (FIG. 1). In the consensus sequence, an asterisk indicates residues conserved in all species. A blackened circle indicates residues that are conserved in most species.
[0394] FIG. 2 shows a human STOP-1 protein. The boxed sequence indicates the signal cleavage site. The Triple Helix Domain is underlined.
Example 2
STOP-1 is Overexpressed in Tumors
[0395] A proprietary database containing gene expression information from microarrays (GeneExpress®, GeneLogic Inc., Gaithersburg, Md.) was analyzed for the expression of STOP-1 mRNA in various tumors (BLIST analysis, proprietary software written an developed at Genentech, Inc. for use with the GeneExpress® database). Some of the types of tissues analyzed included: adipose, adrenal, blood vessel, bone breast, cervix, CNS, colorectal endometrium, esophagus, gall bladder, head and neck, heart, hematopoetic, kidney, liver, lung, lymphoid, muscle, myometrium, euroendocrine, ovary, pancreas, prostate, skin, small intestine, soft tissue, stomach, testis, thymus, thyroid and urinary and normal tissues (breast, colon, lung, ovarian and kidney). STOP-1 mRNA levels were observed to be especially elevated in bone, breast, cervix, colorectal, endometrium, esophagus, glioma, head & neck, kidney, lung, euroendocrine, ovary, pancreas, skin, soft tissue, stomach, thyroid and urinary tumors.
[0396] RNA expression levels were also determined by reverse transcription (RT) and polymerase chain reaction (PCR) amplification of the specific target, STOP-1 (SEQ ID NO:1), in total RNA from a variety of tumors and normal tissues (breast, colon, lung, ovarian and kidney). Tissues were obtained by the Genentech pathology Department and RNA prepared by cesium chloride centrifugation.
[0397] One step RT-PCR amplification reactions consisted of 10× Buffer A (Applied Biosystems), 10 Units RNase inhibitor, 200 uM dATP, dCTP, dGTP, dTTP, 5 mM MgC12, 1.25 Units Taq Gold Polymerase, 25 Units MULV reverse transcriptase (PE Biosystems), 50 ng total RNA, 200 nM gene-specific hybridization probe (FAM-CATCCAGTAGAAGCATCTCCTTTTGGGTAA-TAMRA) (SEQ ID NO:23), 300 nM gene-specific forward primer (GGGTTGGCACTTGTTCAGA) (SEQ ID NO:24) and 300 nM gene-specific reverse primer (CAATAATGATGCGAGAAACTGAAT) (SEQ ID NO:25) in water to a final volume of 50 uL. Thermal cycling conditions were as follows: 1) 48° C., 30 minutes; 2) 95° C., 10 minutes; and 3) 40 cycles of 95° C., 15 seconds and 60° C., 1 minute using an ABI 7700 Sequence Detection System. Transcript levels were normalized to the housekeeping genes GAPDH or RPL19.
[0398] Taqman analysis confirmed the overexpression of STOP-1 mRNA in these tumors, especially pancreas, kidney, breast, lung, ovarian, colorectal tumors, soft tissue, stomach thyroid and urinary tumors. Fourteen out of eighteen breast and colon tumor samples showed five to twenty-seven fold overexpression as compared with normal samples. Other tumor types also showed an increase although to a lesser degree. Those tumor types included adrenal, bone, cervical, endometrium, esophagus, head & neck, kidney, liver and euroendocrine.
Example 3
In Situ Hybridization Studies
[0399] (a) Probe Synthesis [0400] 12.0 l (125 mCi) of [alpha-33P]UTP (NEN/Perkin-Elmer NEG307H) was speed vac dried in a siliconized 1.5 l microfuge (Eppendorf) tube. To each tube having dried 33P-UTP, the following reagents were added and incubated for 1 hour in a 37° C. water bath: [0401] 2.0 l Transcription Optimized 5× Buffer (Promega, P1181) [0402] 2.0 l SQ H2O [0403] 1.0 l DTT, 100 mM (Promega, P1171) [0404] 2.0 l rNTP mix, 2.5 mM [10 l each of 10 mM rATP (Promega, P113B), rCTP (Promega, P114B) & GTP (Promega, P115B)+10 l Nuclease-Free H2O (Promega, P1193)] [0405] 1.0 l RNasin Ribonuclease Inhibitor (Promega, N2511) [0406] 1.0 l DNA Template (1 ug of linear PCR-amplified DNA template encoding a portion of the human STOP-1 coding sequence flanked by the RNA polymerase promoter sequences of bacteriophage T7 on the upper strand for sense control probe transcription and of bacteriophage T3 on the bottom strand for antisense probe transcription)
[0407] The sequence in the transcription reaction with the sense and anti-sense probes was:
TABLE-US-00014 (SEQ ID NO: 26) 5'-GGGAGCCATG CGACCCCAGG GCCCCGCCGC CTCCCCGCAG CGGCTCCGCG GCCTCCTGCT GCTCCTGCTG CTGCAGCTGC CCGCGCCGTC GAGCGCCTCT GAGATCCCCA AGGGGAAGCA AAAGGCGCAG CTCCGGCAGA GGGAGGTGGT GGACCTGTAT AATGGAATGT GCTTACAAGG GCCAGCAGGA GTGCCTGGTC GAGACGGGAG CCCTGGGGCC AATGTTATTC CGGGTACACC TGGGATCCCA GGTCGGGATG GATTCA-3'
[0408] 1.0 l RNA Polymerase T3 (Promega, P2083) for antisense probes or [0409] 1.0 l RNA Polymerase T7 (Promega, P2075) for sense probes.
[0410] Next, 1 l of RQ1 RNase-Free DNase (Promega, M6101) was added to the Eppendorf tube containing the radioactively-labeled RNA probe and the reaction was further incubated for 15 minutes at 37° C. This step degraded the DNA template in the reaction. To stop the degradation reaction, 90 l TE (10 mM Tris pH 7.6/1 mM EDTA pH 8.0) was added to the Eppendorf tube. Unincorporated nucleotides are removed using an RNeasy Mini Kit (Qiagen 74104, Germantown, Md.).
[0411] The probe yield was determined by pipetting onto separate rounds of DE81 ion exchange paper (Whatman, 3658 325) 1 l prefiltered probe and 1 l of probe following filtration. The samples are immersed in 6 ml of Biofluor (Packard, 6NEF-961) in scintillation vials (Wheaton Scientific, 986701) and counted on Beckman LS 6500.
[0412] Next, the probe was analyzed on a 6% Polyacrylamide TBE/Urea gel (Novex EC6865, Invitrogen, Carlsbad, Calif.) to confirm that the transcript was of the proper length. 1×106 cpm of probe or 2 l of Novagen Perfect RNA marker 0.1-1 kb (Novagen 69924-1) was added to 3 l TBE/Urea Sample Buffer (Novex, LC6876). The RNAs were denatured on 95° C. heat blocks for 3 minutes and then immediately chilled on ice. The samples were run at 180-250 volts for 45 minutes. The gel was exposed for 1 hour to Biomax MS film (Kodax, 829 4985) with an intensifying screen in -70° C. freezer.
[0413] (b) In Situ Hybridization
[0414] In situ hybridization analysis was performed initially on sections of normal and tumor tissues. The slides were hybridized to human STOP-1 sense and anti-sense RNA probes using the techniques described below. Further analysis was conducted on sections of tissue microarrays (TMAs) containing numerous normal and tumor tissue specimens. The TMA sections were hybridized exclusively with antisense RNA probes.
[0415] The slides were baked in an oven to adhere tissue to glass at 37° C. overnight followed by 65° C. for 30 minutes. The sections were deparaffinized in a Leica Autostainer XL (Leica, Deerfield, Ill.) by incubating 3 times for 5 minutes each in Xylenes (Richard Allen, Kalamazoo, Mich.) then rehydrating through a graded ethanol series to distilled water. Slides were then washed twice in 2×SSC (0.3 M NaCl, 0.030 M NaCitrate, pH 7.0) for 5 minutes each time. The slides were treated for 15 minutes in a 20 g/ml Proteinase K (Roche Diagnostics, Indianapolis, Ind.) in 10 mM Tris pH 8.0/0.5 M NaCl solution at 37° C. and washed for 10 minutes in 0.5×SSC (0.075 M NaCl, 0.007 M NaCitrate, pH 7.0). The slides were dehydrated with an ethanol gradient (70%-95%-100%) and air-dried. The slides were covered with 100 l hybridization buffer (50% formamide, 10% dextran sulfate, and 2×SSC) and prehybridized for 1-4 hours at 42° C. The [33P]-labeled single-stranded STOP-1 probe (anti-sense orientation) referenced above at a concentration of 2×106 cpm was dissolved in 100 l of hybridization buffer containing 1 mg/ml tRNA and added to the prehybridization buffer on one of the slides, mixed well, covered with coverslip, and allowed to hybridize overnight at 55° C. in a sealed humidified container.
[0416] The foregoing hybridization procedure was performed on another slide from the same tissue block using the same [33P]-labeled single-stranded STOP-1 probe in the sense orientation.
[0417] After hybridization, the slides were washed twice for 10 minutes in 2×SSC containing 1 mM EDTA at room temperature, and then incubated for 30 minutes at 37° C. in 20 g/mL RNase A in 10 mM Tris pH 8, 0.5 M NaCl. The slides were washed for 10 minutes in 2×SSC containing 1 mM EDTA at room temperature, then washed 4 times for 30 minutes each in 0.1×SSC containing 1 mM EDTA at 55° C., and then washed in 0.5×SSC for 10 minutes at room temperature. The slides were dehydrated for 2 minutes each in 50%, 70%, and 90% ethanol containing 0.3M ammonium acetate, and allowed to dry in the air.
[0418] Slides were apposed to X-ray film (Biomax MR Film, Kodax, 870 1302) for 16 hours to obtain a preliminary assessment of the success of the experiment. Slides were then dipped in NTB2 Emulsion (Kodax, 165 4433) [1:1 dilution with H20], allowed to dry overnight in complete darkness, transferred into light tight boxes with desiccant and allowed to expose for 4 weeks. After 4 weeks, slides were developed using D-19 developer [1:1 dilution with H20] for 3 minutes at 15° C., rinsed and fixed in GBX fixer for 6 minutes at 15° C. Slides were counterstained with hematoxylin and eosin prior to examination by a pathologist.
[0419] Table 7 shows the results of the in situ hybridization experiments:
TABLE-US-00015 TABLE 7 NORMAL TISSUE TUMOR TISSUE TISSUE TYPE (# of positive/total) (# of positive/total) Lung 2/16 adult 41/49 adenocarcinoma 27/30 squamous cell carcinoma 1/1 large cell carcinoma 2/5 lung neuroendocrine tumor Cell Lines - all tested negative Calu-6 (lung carcinoma anaplastic) SK-MES cell (squamous cell lung) H322 A549 (lung carcinoma) H522 Breast 0/6 adult 2/14 adult Colon 0/5 adult 4/8 adult Small Intestine 1/5 adult -- Pancreas 0/2 adult 3/5 adult Heart 0/2 fetal -- 0/4 adult -- Placenta 0/5 adult -- Aorta 0/1 adult -- Blood Vessel 0/1 adult -- Thymus 0/1 adult -- Trachea 0/1 adult -- Liver 0/3 adult 2/4 adult hepatocellular carcinoma Thyroid 1/3 adult -- Skin 1/1 adult in follicle and 4/4 adult melanoma dermis Stomach 0/3 adult -- Brain 0/4 adult -- Spleen 0/4 adult -- Lymph Node 0/3 adult 0/4 lymphomas adult Prostate 0/3 adult 0/1 lymphomas adult Ovary 1/1 adult 2/2 adult Urinary Bladder 0/1 adult 3/3 transitional cell carcinomas Gall Bladder 0/1 adult -- Kidney 0/2 adult 0/4 adult Adrenal 0/1 adult -- Endometrium -- 2/3 adult Cartilage -- 0/1 chondrosarcoma Adipose -- 0/1 liposarcoma Fetal Tissues 0/1 aorta, blood vessel, -- thymus, trachea, liver, lung
[0420] The in situ hybridization experiments showed that most normal samples tested negative for STOP-1 mRNA. On the other hand, an astonishingly high number of lung tumors, colon tumors, pancreatic tumors, hepatocellular carcinomas, melanomas, ovarian cancers, endometrial cancers and urinary bladder transitional cell carcinomas showed significant STOP-1 mRNA expression. Furthermore, STOP-1 mRNA was mainly expressed in the stroma (e.g., lung squamous cell carcinomas, adenocarcinomas, breast carcinoma) and not the epithelial compartments of most tumors with the exception of the melanomas which expressed STOP-1 mRNA in the neoplastic cells. The few normal tissues that tested positive for STOP-1 mRNA showed expression in the stromal tissue, with the exception of the thyroid tissue, which expressed STOP-1 mRNA in its epithelial cells. "-" indicates that the tissues were not examined.
Example 4
Northern Analysis of STOP-1 mRNA in Normal Tissues
[0421] Human and mouse STOP-1 cDNA probes (full length coding sequences) were radiolabeled with the random-prime kit (Perkin-Elmer) and applied for analysis of human multiple tissue and mouse embryo blots (Clontech) according to the instructions of the manufacturer. The results can be seen in FIGS. 3a and b.
[0422] In normal adult human tissues, the highest STOP-1 mRNA expression was detected in placenta, heart and skeletal muscle (FIG. 3a). FIG. 3b shows a northern blot that indicates that strong STOP-1 mRNA expression in mouse embryos at 7, 11, 15 and 17 days of development.
Example 5
STOP-1 DNA Constructs
[0423] (a) Mammalian Cell Expression
[0424] All wild type and mutant human, mouse and zebrafish STOP-1 cDNA sequences were made by PCR and expressed in pIRESpuro2 vector (BD Biosiences) or pIRESpuro2 with synthetic 8×HIS tag coding sequence (HIP) for expression in mammalian cells. Human FAP cDNA were expressed in N-terminal pFLAG-CMV vecor (Sigma).
TABLE-US-00016 TABLE 8 Cloning vector/ Construct Forward primer, 5'-3' Reverse primer, 5'-3' 5'/3' cloning sites comments HIP GATCGCGGCCGCACACCACCA GGCCTCACTTAGTGATGGTGA BamHI/NotI pIRESpuro2; TCACCATCACCATCACTAAGT TGGTGATGGTGGTGTGCGGCC HIP-modified GA GC original vector (SEQ ID NO: 27) (SEQ ID NO: 28) with C-terminal HIS, these are 2 adapter oligos zf762HIP CTGCGCTAGCACCATGATGGG GGAAGCGGCCGCTTTTGGAAG NheI/NotI zebrafish TACTAAACTGACTCAACTTT CTCTTCAATGATCA construct (SEQ ID NO: 29) (SEQ ID NO: 30) m762pIRESpuro2 GAAGCTAGCACCATGCACCCC GAAGCGGCCGCTTATTTCGGT NheI/NotI pIRESpuro2 CAAGGCCGCGCGGCCCCCCCG AGTTCTTCAATGATGAT CAGCTGCTGCTCG (SEQ ID NO: 32) (SEQ ID NO: 31) h762pIRESpuro2 CCAGCTAGCACCATGCGACCC CAAGCGGCCGCTTATTTTGGT NheI/NotI pIRESpuro2 CAGGGCCCCGCCGCCT AGTTCTTCAATAATGAT (SEQ ID NO: 33) (SEQ ID NO: 34) h762HIP CCAGCTAGCACCATGCGACCC CAAGCGGCCGCTTTTGGTAGT NheI/NotI HIP CAGGGCCCCGCCGCCT TCTTCAATAATGAT (SEQ ID NO: 33) (SEQ ID NO: 35) ΔTHDh762HIP CCAGCTAGCACCATGCGACCC TCAAAGCTTTCCCTCAGCATT NheI/HindIII h762HIP (1-54, 94-243, CAGGGCCCCGCCGCCT CCATTATACAGGTCCACCACC plus His) (SEQ ID NO: 33) T (SEQ ID NO: 36) ΔΔTHDh762HIP CCAGCTAGCACCATGCGACCC CTCAAAGCTTTCCCTCAGATA NheI/HindIII h762HIP (1-51, 94-243, CAGGGCCCCGCCGCCT CAGGTCCACCACCTCCCTCTG plus His) (SEQ ID NO: 33) (SEQ ID NO: 37) del-N-ter-HIP CCAGCTAGCACCATGCGACCC GGGGAGCTCAGAGGCGCTCGA NheI/SacI HIP (1-32, 54-243, CAGGGCCCCGCCGCCT CGGCGCGGGCA plus His) (SEQ ID NO: 33) (SEQ ID NO: 38) GAAGAGCTCAGGGAAAGCTTT CAAGCGGCCGCTTTTGGTAGT SacI/NotI GAGGAGTCCTGGA TCTTCAATAATGAT (SEQ ID NO: 40) (SEQ ID NO: 35) h762G53AHIP CCAGCTAGCACCATGCGACCC ACAGGTCGACCACCTCCCTCT NehI/SalI HIP CAGGGCCCCGCCGCCT GCCGGAGCT (SEQ ID NO: 33) (SEQ ID NO: 39) GTGGTCGACCTGTATAATGCA CAAGCGGCCGCTTTTGGTAGT SalI/NotI AGGGCCAGCAGGA TCTTCAATAATGAT (SEQ ID NO: 41) (SEQ ID NO: 35) h762N186AHIP CCAGCTAGCACCATGCGACCC AATTGTCGACGCCATTTCAGG NheI/SalI HIP CAGGGCCCCGCCGCCT CTTCCTTGGTCCAA (SEQ ID NO: 33) (SEQ ID NO: 42) TGGCGTCGACAATTAATATTC CAAGCGGCCGCTTTTGGTAGT SalI/NotI ATCGCACTT TCTTCAATAATGAT (SEQ ID NO: 43 (SEQ ID NO: 35) h762C55AHIP CCAGCTAGCACCATGCGACCC ACAGGTCGACCACCTCCCTCT NheI/SalI HIP CAGGGCCCCGCCGCCT GCCGGAGCT (SEQ ID NO: 33) (SEQ ID NO: 39) GTGGTCGACCTGTATAATGGA CAAGCGGCCGCTTTTGGTAGT SalI/NotI ATGGCCTTACAAGGGCCAGCA TCTTCAATAATGAT GGAGTGCCT (SEQ ID NO: 35) (SEQ ID NO: 44 h762C93AHIP CCAGCTAGCACCATGCGACCC CTCAAAGCTTTCCCTCAGAGC NheI/HindIII h762HIP CAGGGCCCCGCCGCCT TTCCCCCTTTTCTCCTTTGAA (SEQ ID NO: 33) T (SEQ ID NO: 45 h762C109AHIP CCAGCTAGCACCATGCGACCC CCATGAGGCCTCTTGTAGTTG NheI/StuI HIP CAGGGCCCCGCCGCCT GGTGTC (SEQ ID NO: 33) (SEQ ID NO: 46 AGCAGGCCTCATGGAGTTCAT CAAGCGGCCGCTTTTGGTAGT StuI/NotIs TGAATTAT TCTTCAATAATGAT (SEQ ID NO: 47 (SEQ ID NO: 35) h762C126AHIP CCAGCTAGCACCATGCGACCC TAAACGTTGCCTCCGCAATTT NheI/AclI HIP CAGGGCCCCGCCGCCT TCCCAAG (SEQ ID NO: 33) (SEQ ID NO: 48 GGCAACGTTTACAAAGATGCG CAAGCGGCCGCTTTTGGTAGT AclI/NotI TTCAAA TCTTCAATAATGAT (SEQ ID NO: 49 (SEQ ID NO: 35) h762C149AHIP CCAGCTAGCACCATGCGACCC GCATGCATTTCTGGCTTTTAG NheI/NsiI h762HIP CAGGGCCCCGCCGCCT CCGAAGTGAGCCA (SEQ ID NO: 33) (SEQ ID NO: 50) h762C153AHIP CCAGCTAGCACCATGCGACCC CTGGCATGCTGCATTTCTGCA NheI/SphI HIP CAGGGCCCCGCCGCCT TTTTA (SEQ ID NO: 33) (SEQ ID NO: 51 GCAGCATGCCAGCGTTGGTAT CAAGCGGCCGCTTTTGGTAGT SphI/NotI TTCACATTCAA TCTTCAATAATGAT (SEQ ID NO: 52 (SEQ ID NO: 35) h762C154AHIP AATGCATGCGCTCAGCGTTGG CAAGCGGCCGCTTTTGGTAGT NsiI/NotI h762HIP TATTTCACA TCTTCAATAATGAT (SEQ ID NO: 53 (SEQ ID NO: 35) h762C166AHIP CCAGCTAGCACCATGCGACCC CCTGAGGCCTCAGCTCCATTG NheI/StuI HIP CAGGGCCCCGCCGCCT AATGTGAAA (SEQ ID NO: 33) (SEQ ID NO: 54 CTGAGGCCTCAGGACCTCTTC CAAGCGGCCGCTTTTGGTAGT StuI/NotI CCATTGAA TCTTCAATAATGAT (SEQ ID NO: 55) (SEQ ID NO: 35) h762C201AHIP CCAGCTAGCACCATGCGACCC TCCGGCGCCAATTCCTTCAGC NheI/KasI HIP CAGGGCCCCGCCGCCT AAGTCCTTCCACAGAAGAAGT (SEQ ID NO: 33) GCGATGAA ATTGGCGCCGGATTAGTGGAT (SEQ ID NO: 56) GTTGCTATCT CAAGCGGCCGCTTTTGGTAGT KasI/NotI (SEQ ID NO: 57 TCTTCAATAATGAT (SEQ ID NO: 35) h762C218AHIP CCAGCTAGCACCATGCGACCC TGAAGCGGTACCAACCCAGAT NheI/KpnI HIP CAGGGCCCCGCCGCCT AGCAACATC (SEQ ID NO: 33) (SEQ ID NO: 58 GGCGGTACCGCTTCAGATTAC CAAGCGGCCGCTTTTGGTAGT KpnI/NotI CCAAAAGGAGA TCTTCAATAATGAT (SEQ ID NO: 59) (SEQ ID NO: 35) h762N52AHIP CCAGCTAGCACCATGCGACCC ACAGGTCGACCACCTCCCTCT NheI/SalI HIP CAGGGCCCCGCCGCCT GCCGGAGCT (SEQ ID NO: 33) (SEQ ID NO: 39) GTGGTCGACCTGTATGCTGGA CAAGCGGCCGCTTTTGGTAGT SalI/NotI AGGGCCAGCA TCTTCAATAATGAT (SEQ ID NO: 60) (SEQ ID NO: 35) h762G53AHIP CCAGCTAGCACCATGCGACCC ACAGGTCGACCACCTCCCTCT NheI/SalI HIP CAGGGCCCCGCCGCCT GCCGGAGCT (SEQ ID NO: 33) (SEQ ID NO: 39) Same as for h762N52AHIO GTGGTCGACCTGTATAATGCA CAAGCGGCCGCTTTTGGTAGT SalI/NotI AGGGCCAGCAGGA TCTTCAATAATGAT (SEQ ID NO: 41) (SEQ ID NO: 35) h762M54AHIP CCAGCTAGCACCATGCGACCC ACAGGTCGACCACCTCCCTCT NheI/SalI HIP CAGGGCCCCGCCGCCT GCCGGAGCT (SEQ ID NO: 33) (SEQ ID NO: 39) Same as for h762n52AHIP GTGGTCGACCTGTATAATGGA CAAGCGGCCGCTTTTGGTAGT SalI/NotI GCGTGCTTACAAGGGCCAGCA TCTTCAATAATGAT GGAGT (SEQ ID NO: 35) (SEQ ID NO: 61) h762P63A, CCAGCTAGCACCATGCGACCC TCGGCCGGCCACTCCTGCTGG NheI/NgoMIV HIP P69AHIP CAGGGCCCCGCCGCCT CCCTTGTAA (SEQ ID NO: 33) (SEQ ID NO: 62) GTGGCCGGCCGAGACGGGAGC CAAGCGGCCGCTTTTGGTAGT NgoMIV/NotI GCTGGGGCCAATGGCATTCCG TCTTCAATAATGAT GGTA (SEQ ID NO: 35) (SEQ ID NO: 63 h762P75A, CCAGCTAGCACCATGCGACCC TGTGCCGGCAATGCCATTGGC NheI/NgoMIV HIP P78A, P81AHIP CAGGGCCCCGCCGCCT CCCCAGG (SEQ ID NO: 33) (SEQ ID NO: 64 ATTGCCGGCACAGCTGGGATC CAAGCGGCCGCTTTTGGTAGT NgoMIV/NotI GCAGGTCGGGATGGATTCAAA TCTTCAATAATGAT GGAGAAAA (SEQ ID NO: 35) (SEQ ID NO: 65) k762K87A, CCAGCTAGCACCATGCGACCC TTCGCCGGCGAATCCATCCCG NheI/NgoMIV HIP K90AHIP CAGGGCCCCGCCGCCT ACCTGGGAT (SEQ ID NO: 33) (SEQ ID NO: 66) TTCGCCGGCGAAGCGGGGGAA CAAGCGGCCGCTTTTGGTAGT NgoMIV/NotI TGTCTGAGGGAAAGCTTT TCTTCAATAATGAT (SEQ ID NO: 67 (SEQ ID NO: 35) M762HIP GAAGCTAGCACCATGCACCCC GAAGCGGCCGCTTTCGGTAGT NheI/NotI HIP CAAGGCCGCGCGGCCCCCCCG TCTTCAATGAT CAGCTGCTGCTCG (SEQ ID NO: 68) (SEQ ID NO: 31) hFAPCMV GAAGCGGCCGCAAGACTTGGG GAAAGATCTCTAGTCTGACAA NotI/BglII N-TER-PfLAG cmv TAAAAATCGTATTT AGAGAAACACTGCTTTAGGA (SEQ ID NO: 69) (SEQ ID NO: 70)
[0425] (b) Baculovirus Expression
[0426] For expression in baculovirus, the following human STOP-1 constructs were made: S31humanSTOP-1-pAcGP67B encoding S31-K243, L94humanSTOP-1-pAcGP67B encoding L94-K243, and E89humanSTOP-1-pAcGP67B encoding E89-K243 of human STOP-1. S31humanSTOP-1-pAcGP67B was generated in a two step PCR approach. The 5' piece was generated from pAcGP67B using primers #161344 (GGATCGTCGGTTTTGTACAATATGT) (SEQ ID NO:71) and #161347 (GGGGATCTCAGACGCAAAGGCAGAATGCGC) (SEQ ID NO:72). The 3' piece was generated from DNA #84694 using primers #161348 (TCTGCCTTTGCGTCTGAGATCCCCAAGGGG) (SEQ ID NO:73) and #161732 (CCGTTCTGCAGTTAATGATGATGATGATGATGATGATGG) (SEQ ID NO:74). The full length insert (which is then subcloned into pAcGP67B) was generated from a PCR reaction of equal parts 5' and 3' pieces using primers #161344 and #161732.
[0427] L94humanSTOP-1-pAcGP67B was generated in a two step PCR approach. The 5' piece was generated from pAcGP67B using primers #161344 and #161349 (GCTTTCCCTCAGCGCAAAGGCAGAATGCGC) (SEQ ID NO:75). The 3' piece was generated from DNA #84694 using primers #161350 (TCTGCCTTTGCGCTGAGGGAAAGCTTTGAGG) (SEQ ID NO:76) and #161346 (CCGGGATCCTTAATGATGATGATGATGATGAT) (SEQ ID NO:77). The full length insert (which is then subcloned into pAcGP67B) was generated from a PCR reaction of equal parts 5' and 3' pieces using primers #161344 and #161346.
[0428] DNA #84694 was amplified by PCR to generate two fragments containing S31-K243 and L94-K243 with C-terminal His tags. PCR fragments were subcloned into the baculovirus transfer vector pAcGP67B (PharMingen), which was then co-transfected with BaculoGold DNA (PharMingen) into Sf9 cells. Recombinant virus was isolated and amplified in Sf9 cells.
[0429] E89humanSTOP-1-pAcGP67B was generated in a two step PCR approach. The 5' piece was generated from pAcGP67B using primers #161344 (GGATCGTCGGTTTTGTACAATATGT) (SEQ ID NO:71) and #161351 (ATTCCCCCTTTTCCGCAAAGGCAGAATGCGC) (SEQ ID NO:78). The 3' piece was generated from DNA #84694 using primers #161352 (TCTGCCTTTGCGGAAAAGGGGGAATGTCTGAG) (SEQ ID NO:79) and #161346 (CCGGGATCCTTAATGATGATGATGATGATGAT) (SEQ ID NO:77). The full length insert (which is then subcloned into pAcGP67B) was generated from a PCR reaction of equal parts 5' and 3' pieces using primers #161344 and #161346.
Example 6
Expression from STOP-1 DNA Constructs and Purification of Proteins
[0430] DNA encoding STOP-1 DNA constructs were transfected into CHO-DP12 cells, CHO-psgb cells (hamster galactosyltransferase I deficient epithelial pgsB-618 CHO cells) (ATCC# CRL-2241) or 293 cells (Roche) using calcium phosphate or fugene 6 transfection reagent according to the instructions of the manufacturer (Roche). The growth medias were supplemented with 1 mM NiC1, 5 mM CaCl2 and 50 mM Tris pH 7.6-8.0. Sixteen hours post transfection the serum containing the media was replaced with serum free media and protein was allowed to accumulate for 4-6 days. The secreted proteins were purified using Ni-NTA agarose beads.
[0431] Proteins from cell lysates were prepared by lysing the cells four days after transfection in buffer containing 50 mM HEPES (pH 7.5), 150 mM NaCl, 1% Triton X-100, 5 mM EDTA, 1× protease inhibitor cocktail (ROCHE, [lysis buffer]). Immunoprecipitation was performed by preclearing cell lysates (2 hr at 4° C.) with 25 l of immobilized protein A/G-agarose beads (Pierce) followed by incubation (4 hr at 4° C.) of aliquots (0.5 ml) of lysates (4×105 cells per aliquot) with anti-HIS epitope antibody (Qiagen) and with protein A/G-agarose beads. Immunoprecipitates were washed three times with lysis buffer and once with phosphate-buffered saline, fractionated by 10% SDS-PAGE, and transferred to a nitrocellulose membrane (Invitrogen). Western blot analysis was performed with mouse anti-HIS and the ECL kit (Amersham).
[0432] FIG. 4a shows that CHO-DP12 cells, used routinely for expression of recombinant proteins, produced little secreted STOP-1. In contrast, a CHO mutant cell deficient in proteoglycan synthesis, CHO-Psgb, produced much more secreted STOP-1 and yielded a better recovery of the secreted proteins in soluble form (FIG. 4b). pRK5 was transfected as a negative control. FIGS. 5a and b show that His-tagged protein can be detected with anti-H is antibodies in cell supernatants and in cell lysates, respectively, of CHO-psgB transfected cells.
[0433] For protein production from baculovirus, Hi5 cells were infected with amplified baculovirus. After 3 days in culture at 27° C., the medium was harvested by centrifugation. The supernatant was supplemented with 50 mM Tris 8.0, 1 mM NiCl2, 5 mM CaCl2, and the pH was adjusted to 7.6. The medium was filtered and loaded onto a Ni-NTA agarose column (Qiagen). The column was washed with 50 mM Tris 8.0, 300 mM NaCl, 2 mM benzamidine, 0.5 mM PMSF, and 5 mM imidazole. Elution was performed in the same buffer with 300 mM imidazole. Fractions containing STOP-1 were pooled and concentrated. Protein was purified over a Superdex-75 column into 50 mM Tris 8.0, 100 mM NaCl, 0.5 mM PMSF, and 2 mM benzamidine STOP-1 containing fractions were pooled, concentrated, and utilized for crystallography trials and other studies.
Example 7
STOP-1 Oligomerization
[0434] Full length STOP-1-HIS proteins or various truncations thereof were expressed in SF9 baculovirus-infected cells or CHO cells as described above. The proteins were purified from the media by using Ni-NTA agarose beads as discussed above. The baculoviral-expressed proteins S31-K243-His, E89-K243-His, or L94-K243-His were loaded onto an 8 mm×300 mm Shodex KW802.5 size exclusion column and eluted with 100 mM NH4HCO3, 200 mM NH4Cl pH7.8 at a flow rate of 1 ml/ml. CHO-psgb expressed full length proteins were loaded onto an 8 mm×300 mm Shodex KW804 size exclusion column and eluted with 25 mM sodium phosphate, 500 mM NaCl at a flow rate of 1 ml/ml. CHO-psgb expressed proteins, M1-M54 fused to L94-K243-His, were loaded onto an 8 mm×300 mm Shodex KW802.5 size exclusion column and eluted with 25 mM sodium phosphate, 500 mM NaCl at a flow rate of 1 ml/ml.
[0435] The eluted protein was analyzed by an Agilent Model 1100 HPLC system with an Agilent multi wavelength detector (UV) connected to a Wyatt MiniDAWN laser light scattering (LS) instrument and a Wyatt Optilab differential refractometer (RI). The average molecular weight of the proteins or their aggregates in each peak was determined by selecting a peak and using the Zimm fitting method (Phillip J. Wyatt, (1993) Analytica Chimica Acta 272:1-40) with the Astra software package (Wyatt, USA). The percentage of aggregation was calculated from the integrated peak areas of the UV signal at 214 nm. See FIG. 6 and FIG. 7 for results.
[0436] Light scatter analysis indicates that the baculovirus S31-K243-His proteins primarily formed trimeric molecules (apparent molecular weight 74 kD, 95% of total integrated peak area) (FIG. 6A). The baculovirus E89-K243-His proteins formed complexes that were approximately 36% trimeric and 64% hexameric (i.e., apparent MW of 126 kD representing 36% of peak area, apparent MW of 61 kD representing 64% of peak area) (FIG. 6B). However, it is believed that the hexameric complexes formed by E89-K243-His are an anomaly due its odd number of cysteines. The baculovirus L94-243 protein, which lacks the triple helix domain, forms trimeric complexes (apparent MW of 59 kD representing 98% of peak area) (FIG. 6c). This indicates that the N-terminal domain of the protein and the triple helix domain are not required for trimerization. The L94-K243 region is sufficient. The THD can act to stabilize the trimers.
[0437] Proteins expressed in CHO-psgb also formed complexes (FIG. 7A-C). Light scatter analysis indicated that the full length-expressed protein (M1-K243-His) formed complexes that were approximately 58% hexameric and 42% trimeric (FIG. 7A). The complexes formed by delta-THD expression (M1-M54 and L94-L243-His) were in part trimeric (apparent MW of 66 kD representing 61% of peak area) and in part heterogeneous in mass (average MW of 627 kD) probably due to the varied glycosylation state of the proteins in CHO cells (FIG. 7B).
[0438] On SDS gels, purified full-length STOP-1 expressed in CHO-psgB cells migrated as a monomer under reducing conditions and as a dimer under non-reducing conditions (without DTT). See FIG. 7c. The results indicate that disulfide bonding can occur between two monomers.
Example 8
Deletion and Point Mutational Analysis of STOP-1
[0439] Full-length STOP-1 (WT)-His, delta-THD-His, delta-delta-THD-His, delta-N-terminus and several point mutation mutants were expressed in CHO-psgb cells as described above. Whole cell extract extracts were prepared. Aliquots from the whole cell extracts or the media in which the cells were cultured were run on an SDS gel under reducing and non-reducing conditions and western blotted with anti-His antibody (ECL detection kit, Amersham).
[0440] FIGS. 8A and B indicates that a disulfide bond can form between two monomers in the THD region in extracts from supernatants and lysates, respectively. Each deletion mutants lacking the THD, which includes cysteine 55 failed to form dimers under non-reducing conditions. Homodimers were present in the supernatant and lysates of cells expressing full length protein under non-reducing conditions, but not under reducing conditions. Table 9 summarizes the results as follows:
TABLE-US-00017 TABLE 9 Construct Secretion Dimerization WT + + delta-THD + - delta-delta-THD + - delta-N-ter + + "+" indicates that the level of secretion or dimerization was the same as wild-type STOP-1. "-" indicates that dimerization was not detectable in this assay.
[0441] Point mutants of human STOP-1 were tested for the ability to secrete and dimerize. FIGS. 9A and B shows that wild-type STOP-1 protein and a G53A mutant were secreted and that homodimers are observed in the in supernatants and lysates. However, a mutant at 186 ("N" mutated to "A"), a potential glycosylation site, was not present in the culture media and did not homodimerize. Many other mutants were also tested. Table 10 summarizes the results as follows:
TABLE-US-00018 TABLE 10 Construct Secretion Dimerization WT + + N52A + + G53A ++ + M54A + + P63A, P69A + + P75A, P78A, P81A + + K87A, K90A + + N186A - - "+" indicates that the level of secretion or dimerization was the same or better ("++") than wild-type STOP-1. "-" indicates that secretion or dimerization was not detectable in this assay. All these mutants except N186A were secreted and ran similar to WT on SDS gels.
[0442] FIGS. 10A and B show the state of the a human STOP-1 protein having point mutations at cysteine residues 55, 93 and 109. Point mutations were also made at other cysteines throughout the protein. Table 11 summarizes the results as follows:
TABLE-US-00019 TABLE 11 Construct Secretion Dimerization WT + + C55A + - C93A + + C109A - C126A - C153A - C154A - C153A, C154A -/+ -/+ C166A - C201A - C218A - "+" indicates that the level of secretion or dimerization was the same as wild-type STOP-1. "-" indicates that dimerization was not visibly detectable in this assay. "-/+" indicates that secretion or dimerization was weakly detectable in this assay.
[0443] All of the mutants were expressed, but only two were secreted--C55A and C93A. C93A was secreted and formed dimers in contrast to C55A, which was secreted but did not form dimers indicating that C55 is required for intra-subunit disufide bonding.
Example 9
Wnt Pathway Upregulates STOP-1 mRNA
[0444] The STOP-1 gene is located on human chromosome 8 between 8q22 and 8q23. The gene is located close to genes encoding proteins important in the Wnt signalling pathway such as the FZD6 gene (frizzled homolog 8 (drosophila)), the WISP-1 gene (WNT1 inducible secreted protein 1). There are at least three regulatory genes in the Wnt pathway that are mutated in primary human cancers and experimental tumors of other species.
[0445] MMTV-Wnt-1 transgenic mice were prepared by Genentech, Inc. These mice overexpress the Wnt-1 protein under the control of the MMTV promoter. The C57Mg cells do not overexpress Wnt-1. Breast tumors in these mice were removed. mRNA was extracted from the breast tumors or from C57Mg mammary epithelial cells. A reverse transcriptase reaction was performed with an oligo(dT) primer, the avian myeloblastosis virus reverse transcriptase (Promega) and the extracted mRNA according to manufacturer's instructions (Promega). PCR on the reverse transcribed products was carried out with Ex-taq polymerase (Takara) using mRLP19 primers and mouse STOP-1 primers:
TABLE-US-00020 mRPL19: 5'-ATCGCCAATGCCAACTCCCGTCA-3' (SEQ ID NO: 80) and 5'-GCTTGCGTGCTTCCTTGGTCTTA-3'. (SEQ ID NO: 81)
[0446] mRLP19 is the murine mitochondrial ribosomal protein L19 (MRPL19), a housekeeping protein. PCR using primers against mRLP19 was used as a control for the extraction and PCR of mRNA.
TABLE-US-00021 mSTOP-1: 5-TGCTGCTGCAGCTGCCCGCGCCGTCGAG-3 (SEQ ID NO: 82) and 5-TCCAGTAGAAGCATCTCCTTTTGGGTAA-3. (SEQ ID NO: 83)
[0447] The results show that mSTOP-1 mRNA is expressed in the breast tumors of MMTV-Wnt-1 transgenic mice (FIG. 11, lanes T1-T7), whereas it is not expressed in C57Mg normal mouse mammary epithelial cells ("N"). Thus, a connection is suggested between the Wnt signalling pathway and STOP-1 expression.
Example 10
Coexpression of STOP-1 with Other Genes
[0448] A proprietary database containing gene expression information (GeneExpress®, GeneLogic Inc., Gaithersburg, Md.) was analyzed for genes that are expressed in the same tissues as STOP-1 (BLIST analysis, proprietary software written an developed at Genentech, Inc. for use with the GeneExpress® database). By this method, several genes having significant correlation of expression in breast and colon tumors were identified. Genes that were coexpressed in breast and colon tumors included, Wisp-1 (a WNT target gene), SFRP2 (a soluble WNT receptor), fibroblast activation protein (FAP), a cell surface serine protease that has been implicated in ion cancer), collagen type I alpha 2 chain, collagen type V alpha 2 chain, Thrombospondin 2 (THBS2) (ECM), ADAM12 (a MMP enzyme), OB-Cadherin and OSF-2 (TCI protein). The later genes suggest involvement of STOP-1 in formation and/or modulation of the extracellular matrix.
Example 11
STOP-1 is Cleaved by MMP-7 and MMP-9 In Vitro
[0449] Materials: Human his-tagged STOP-1 protein was produced using a baculovirus expression system. Matrix metalloproteases (MMPs)-1, -2, -3, -7, and -9 were purchased from Enzyme Systems Products. Trypsin was obtained from Sigma.
[0450] Proteolytic Digestion of STOP-1: Prior to reaction with STOP-1, MMPs were activated with 1 mM p-aminomercuric acetate for 1 h at 37 degrees Celsius. STOP-1 (3 ng) was digested with proteases (50 or 250 nM) in a final volume of 20 μl for 4 hours at 37 degrees Celsius. Buffer A (50 mM Tris, pH 87.5, 10 mM CaCl2, 10 nM ZnCl2 and 100 mM NaCl) was the buffer used for the MMP cleavage reaction. Buffer A lacking ZnCl2 was the buffer used for the trypsin cleavage reactions. The MMP reactions were terminated by addition of EDTA (15 mM). The trypsin reaction was terminated by addition of PMSF (1 mM). Samples were then analyzed by SDS-PAGE and Coomassie staining.
[0451] FIG. 23 shows the cleavage of baculoviral expressed human STOP-1 protein by various proteases in vitro. MMP-7 also cleaved human STOP-1 in vitro, whereas MMP-1, -2 and -3 produced minimal or no cleavage products (data not shown). MMP-7 produced STOP-1 cleavage products of about 23 and 21 kDa. In contrast, MMP-9 produced STOP-1 cleavage products of about 22 and 18 kDa. Tyrpsin produced 20 and 22 kDa fragments (data not shown). These data suggest that STOP-1 activity may be regulated by proteolysis. This can be particularly relevant with tumor stromal-associated proteases such as MMP-7 and MMP-9.
Example 12
STOP-1 Expression Promotes 3T3 Proliferation
[0452] (a) Cell Culture and Generation of 3T3 STOP-1 Stable Cell Lines
[0453] 3T3 cells were maintained in DMEM supplemented with 10% FCS. Mouse and human STOP-1-expressing cell lines were established by transfection of 3T3 cells with m762pIRESpuro2 and h762pIRESpuro2 vectors using FuGENE 6 transfection reagent (Roche) according to the instructions of the manufacturer. STOP-1 clones were established by selecting transfected cells with puromycin (4 g/ml).
[0454] (b) In Vitro Proliferation Assay
[0455] 3T3 cells were transfected with mouse STOP-1, human STOP-1 or vector alone as described above and were plated in 96-well plates at 1.5×103 cells in DMEM with 10% FCS. Twelve hours later the media was change to DMEM with 1.5% FCS and 10 uCi/ml [3H]-thymidine. After 12 hours, 48 hours and 96 hours, the cells were harvested onto a GF/C filter using Packard's 96-well Filtermate 196, washed and counted on a top count, microplate scintillation counter (Packard). The results show that clones expressing STOP-1 demonstrated increased proliferation as compared with vector alone-transfected clones (puro2 and phi). See FIGS. 12A and B.
[0456] (c) Retroviral Expression in 3T3 Cell Lines
[0457] A human full-length STOP-1 cDNA was cloned from H762pirespuro2 into the pMSCV1(puro) vector (Clontech) and introduced into 3T3 fibroblasts and 293 cells by retroviral infection using the method described by Maecker et al., (Maeker H. L., et al., Cancer Cell 2, 139-148, 2002). Briefly, 5000 cells/well were plated in 96 well plates and switched to low serum media (0.25% fetal calf serum) the following day. The retrovirally infected cells were selected in puromycin (3 ug/ml). After 24 hours, cell proliferation was measured using the Cell Titer Kit (Promega). FIG. 13C shows that infection with STOP-1 retrovirus promotes proliferation of both 3T3 and 293 cells, respectively, as compared to 3T3 or 293 cells infected with a control.
[0458] For detection of STOP-1 protein, whole cell lysates of cell pellets (˜2×106 cells) were prepared and incubated with antibody S7-IgG (1 μg) described below and subsequently immunoprecipitated with protein A/G Immunoprecipitates were then denatured and transferred to nitrocellulose membranes. STOP-1 was then detected using rabbit polyclonal anti-STOP-1 antibodies. FIGS. 13A and B show expression of STOP-1 in 3T3 and 293 cells in whole cell lysates following infection with STOP-1 retrovirus as compared to a vector control, Babe.
Example 13
STOP-1 Expression Causes Tumorigenesis in mice
[0459] Female athymic nu/nu mice (Charles River Laboratory, [5 animals per group]) were inoculated subcutaneously with 1×106 3T3 cells stably transfected with mouse or human STOP-1, RAS cDNA (Hudziak R M, Lewis G D, Shalaby M R, Eessalu T E, Aggarwal B B, Ullrich A, Shepard H M. (1988) Proc Natl Acad. Sci. 85(14), pp.:5102-6) or an empty puro2 vector (p2). Tumor growth was measured 1 time per week. The stably 3T3 cell lines were also evaluated for STOP-1 expression by preparing whole cell lysates of pelleted cells, performing SDS-PAGE on aliquots of the lysates or cell culture media, and western blotting the gels and probing them with rabbit anti-STOP-1 antibody. Cells transfected with RAS cDNA served as positive controls. See FIG. 14A-C.
[0460] The results show that murine STOP-1 was present in the lysates of all of the NIH 3T3 clones, but was only present in the cell culture media of two of the three NIH 3T3 clones (FIGS. 14B and C, respectively). In other words, clone 18 expressed the protein intracellularly but was defective at secreting the murine STOP-1 protein. See FIG. 14B. Further, the two clones that secreted STOP-1 produced tumors in nude mice whereas the clone that was deficient in secretion did not (FIG. 14A). These results suggest that secreted murine STOP-1 by itself can be tumorigenic whereas intracellularly expressed STOP-1 is not.
[0461] The results also show that human STOP-1 can drive tumorigenesis in mice. See FIG. 15. Tumors grew in several mice that expressed the human STOP-1 protein and in the RAS control, but not in the mice treated with cells transfected with vector alone. Like the mouse STOP-1, these results suggest that secreted human STOP-1 by itself can be tumorigenic.
Example 14
STOP-1 Promotes Wound Healing
[0462] Malignant melanoma SK-MEL-31 cells were maintained in DMEM supplemented with 10% FCS. SK-MeI-31 cells were cultured in 6 well dishes (Corning) until they reached subconfluency, then starved for 8 hours in DMEM with 2% heat-inactivated FCS. The cells were then treated for 2 hours in FCS-free DMEM with 10 g/ml mitomycin C (Sigma) and subjected to the following in vitro wound closure assay.
[0463] A cell-free area was introduced by scraping the SK-MEL-31 cell monolayer with a yellow pipette tip. Cell migration to the cell-free area for 48 hours after scraping was evaluated while the cells were maintained in DMEM supplemented with 2% heat-inactivated FCS in the absence or presence of 30 ng/ml EGF (Roche), 1 g/ml of full length baculovirus expressed human STOP-1 protein, both or none. FIGS. 16A-E are phase-contrast photographs of the scraped area 48 hours after scraping.
[0464] The results show that cells migrated to the scraped area when treated with EGF or STOP-1, especially STOP-1 expressed from CHO-psgB cells. The results indicate that STOP-1 promotes cell migration. Cell migration occurs in tumor growth as it invades surrounding tissue. The data further supports the tumor promoting properties of STOP-1.
Example 15
Monoclonal Antibody Development
[0465] Ten BALB/c mice (Charles River Laboratories, Wilmington, Del.) were hyperimmunized with recombinant polyhistidine-tagged (HIS8) human STOP-1 (a.k.a. DNA 145960) transiently expressed in PSGB chinese hamster ovary cells (Genentech, Inc., South San Francisco, Calif.) in Ribi adjuvant (Ribi Immunochem Research, Inc., Hamilton, Mo.). B-cells from five mice demonstrating anti-STOP-1 antibody titers were fused with mouse myeloma cells (X63.Ag8.653; American Type Culture Collection, Rockville, Md.) using a modified protocol analogous to one previously described (Kohler, G. and Milstein, C. (1975) Nature 256: 495-497; Hongo, J. S., et al., (1995) Hybridoma 14:253-260).
[0466] After 10-14 days, the supernatants were harvested and screened for antibody production by direct enzyme-linked immunosorbent assay (ELISA). One positive clone (6B12.1.7), showing the highest immunobinding after the second round of subcloning by limiting dilution, was injected into Pristane-primed mice (Freund YR and Blair PB (1982) J Immunol 129:2826-2830) for in vivo production of MAb. The ascites fluid was pooled and purified by Protein A affinity chromatography (Pharmacia fast protein liquid chromatography [FPLC]; Pharmacia, Uppsala, Sweden) as previously described (Hongo et al., 1995, supra). The purified antibody preparation was sterile filtered (0.2-m pore size; Nalgene, Rochester N.Y.) and stored at 4° C. in phosphate buffered saline (PBS). The hybridoma clone producing the 6B12 antibody was deposited under the terms of the Budapest Treaty with the American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, Va. 20110-2209, USA on Mar. 28, 2003 as "6B12.1.7".
[0467] The binding site for the 6B12 antibody was mapped to an N-terminal region of human STOP-1. The His-tagged constructs encoding the human full length protein, the delta-THD protein, the delta-delta-THD protein, the delta-N-terminal protein and the zebrafish full length protein were expressed in CHO-psgB cells. Aliquots of transfected cell extracts were run on SDS-PAGE, western blotted and probed with either anti-His antibody or 6B12 antibody.
[0468] The 6B12 monoclonal antibody worked well on westerns. See FIG. 17C. It bound to all of the human proteins expressed as described above except for the delta-N-terminal protein. Therefore, the binding epitope of the 6B12 antibody maps to the N-terminal amino acids #33-52 of human STOP-1. The 6B12 antibody did not recognize the zebrafish STOP-1 protein on a western blot.
Example 16
Phage-Derived Antibodies Against STOP-1
[0469] Overview: Phage-derived antibodies against STOP-1 were made using, in part, materials and methods described in U.S. Provisional Application No. 60/385,338 ("the '338 application"), filed Jun. 3, 2002. In this study, phagemids were further modified and the resultant antibodies were screened based on binding to oligomerized STOP-1.
[0470] Construction of Anti-Her2 Fab and F(ab)'2 phagemid: The phagemid vector, pS0643 (also known as phGHam-g3, e.g., U.S. Pat. No. 5,688,666, Example 8), contains pBR322 and f1 origins of replication, an ampicillin resistant gene, an E. coli alkaline phosphatase (phoA) promoter (Bass et al., (1990) Proteins 8:309-314), and a sequence encoding a stII secretion signal sequence fused to residues 1-191 of human growth hormone (hGH) and a sequence encoding the C-terminal residues 267-421 of protein III of M13 phage (hereinafter, cP3). The pS0643 phagemid also contains an XbaI site and an amber stop codon following residue 191 of hGH. The stII secretion signal sequence can export a protein to the periplasm of a bacteria cell (e.g., a light chain region (LC) of an antibody). In this study, the sequence encoding the human growth hormone (hGH) was removed from the pS0643 vector and replaced with a NsiI/XbaI nucleic acid fragment encoding a humanized anti-Her2 Fab fragment ("h4D5" sequence) ligated in frame with the stII secretion signal (humAb4D5-8, see Carter et al., (1992) PNAS 89:4285-4289, Table 1 and FIG. 1 therein or U.S. Pat. No. 5,821,337, for sequence).
[0471] The h4D5 antibody is a humanized antibody that specifically recognizes a cancer-associated antigen known as Her-2 (erbB2). In this study, the h4d5 was obtained by polymerase chain reaction using the humAb4D5 version 8 ("humAb4D5-8") sequence and primers engineered to give rise to a 5' NsiI site and a 3' XbaI site in the PCR product (Carter et al., (1992) PNAS 89:4285-4289). The PCR product was cleaved with NsiI and XbaI and ligated into the pS0643 phagemid vector. The h4D5 nucleic sequence encodes modified CDR regions from a mouse monoclonal antibody specific for Her-2 in a mostly human consensus sequence Fab framework. Specifically, the sequence contains a kappa light chain (LC region) upstream of VH and CH1 domains (HC region). The method of making the anti-Her-2 antibody and the identity of the variable domain sequences are provided in U.S. Pat. Nos. 5,821,337 and 6,054,297.
[0472] The pS0643 plasmid containing humanized 4D5 (version 8) was still further modified. For example, a herpes simplex virus type 1 glycoprotein D epitope tag (gD tag) was added in frame to the c-terminus of the LC using site-directed mutagenesis. Following the stop codon downstream of the LC, a ribosome binding site and nucleic acid molecule encoding a stII signal sequence were ligated to the N-terminus of the HC sequence. Consequently, the HC sequence is in frame with the C-terminal domain of the p3 (cP3), a minor coat protein of M13 phage. Thus, a Fab displayed on phage can be produced from one construct. This Fab phagemid vector is referred to as pV0350-2b (FIGS. 25A-H) and can be schematically illustrated as FIG. 24A.
[0473] To generate F(ab)'2 displayed on phage, the pV0350-2b vector was further modified by inserting a dimerizable leucine zipper GCN4 sequence (GRMKQLEDKVEELLSKNYHLENEVARLKKLVGERG) (SEQ ID NO:84) between the HC and cP3 sequences by cassette mutagenesis. The GCN4 leucine zipper brings two sets of LC/HC-cP3 fusion polypeptides together in the E. coli periplasm and presents the dimer on the surface of phage. This F(ab)'2 phagemid vector is referred to as pV0350-4 (FIGS. 26A-H) and can be schematically illustrated as FIG. 24B.
[0474] Generating F(ab) Libraries with H1/H2/H3 Diversity for Use in STOP-1 Selection:
[0475] A diversified library for finding anti-STOP-1 antibodies was created by mutating the sequences encoding the HC variable regions in the pV0350-2b and pV0350-4 vector using Kunkel mutagenesis and screening the phage containing them based on binding to human STOP-1 by ELISA assay. The screening method is described in greater detail below. Other antibodies having greater specificity or affinity to STOP-1 can be obtained by, e.g., further mutagenizing the Fab and F(ab)'2 sequences (e.g., by Kunkel mutagenesis, (Kunkel et al., (1987) Methods Enzymol. 154:367-382)) in their LC CDR regions and screening them by binding to STOP-1.
[0476] Expression of phage: E. coli strain SS320 was transformed with the mutagenized DNA described above by electroporation. The size of the libraries was approximately 109. Transformed bacterial cells were grown up overnight in the presence of helper phage KO7 to produce displaying phage that could still infect other bacterial cells. Next, E. coli strain XL-1 Blue (Strategene, San Diego, Calif.) was infected with F(ab) or F(ab)'2 phage and then K07 helper phage (Strategene, San Diego, Calif.) were grown in 2YT media at 37° C. for 20 hours and phage was harvested as described (Sidhu et al., Methods Enzymol. (2000), 328:333-363). Briefly, phage was purified by first precipitating them from the overnight culture media with polyethylene glycol, and resuspended in PBS. Phage were quantitated by spectrophotometer with its reading at 268 nm (1 OD=1.13×1013/ml).
[0477] Phage sorting: The phage libraries were subjected to four rounds of sorting. 96-well Nunc Maxisorp plates were coated with 100 ul/well of target antigen (CHO-psgb-expressed human his-tagged STOP-1 full length or baculovirally-expressed human his-tagged #94-243 amino acids ("short form")) (5 ug/ml) in PBS at 4° C. overnight or room temperature for 2 hours. The plates were blocked with 65 ul 1% blocking protein for 30 min and 40 ul 1% Tween20 for another 30 min (blocking protein: 1st round--bovine serum albumin (BSA), 2nd round--ovalbumin, 3rd round--casein, 4th round--ovalbumin) Next, the library phage was diluted to 3˜5 O.D/ml with 1% BSA with 0.1% Tween 20 (1 O.D=1.13×1013 phage/ml). In general, the phage input was 1st round 3˜5 O.D/ml, 2nd round 3 O.D/ml, 3rd round 0.5˜1 O.D/ml and 4th round input 0.1˜0.5 O.D/ml. The diluted phage was incubated for 30 minutes at room temperature. The wells were washed at least five times continuously with PBS and 0.05% Tween 20. The blocked library phage was added 100 ul/well to 8 target antigen-coated wells and 2 uncoated wells at room temperature for 1 hour. The plates were washed continuously at least 10 times with PBS and 0.05% Tween 20. The phages were eluted with 100 ul/well of 100 mM HCl at room temperature for 20 minutes. The eluted phages (from coated wells) and background phage (from uncoated wells) were collected in separate tubes. The eluted collections were neutralized by adding 1/10 volume 1M Tris pH 11.0 to both tubes. BSA was added to a final 0.1% into the tube of eluted phage. The eluted phage was heated at 62 C for 20 minutes. To titer the phage, 90 ul of log phase XL-1 (OD 600 nm˜0.1-0.3) was infected with 10 ul eluted phage or background phage at 37° C. for 30 minutes. Next, the infected cells were serially diluted in 10 fold increments with 90 ul 2YT. 10 ul aliquots of the infected cells were plated per carbenicillin plate.
[0478] To propagate the phage, approximately 400 ul of eluted phage was used to infect ˜4 ml log phase XL-lsoup (OD 600 nm˜0.1-0.3) at 37° C. for 30-45 minutes. Helper phage, KO7, and carbenicillin were added to the infection at a final concentration of 1×1010 pfu/ml KO7 and 50 ug/ml cabenicillin at 37° C. for another hour. The culture was grown in 50:50 2YT/CRAP media with carbenicillin 50 ug/ml and 50 ug/ml kanamycin to final volumes of 20˜25 ml at 37° C. overnight (or at least 17 hours). The next day, the culture was grown at 30 C for another 2 hours to increase the phage yield.
[0479] The phage was purified by spinning down the cells at 8000 rpm for 10 minutes. The supernatant was collected. 20% PEG/2.5M NaCl was added at 1/5 of the supernatant volume, mixed and allowed to sit on ice for 5 minutes. The phage was spun down into a pellet at 12000 rpm for 15 minutes. The supernatant was collected and spun again for 5 minutes at 5000 rpm. The pellets were resuspended in 1 ml PBS and spun down at 12000 rpm for 15 minutes to clear debris. The steps starting with the PEG/NaCl addition were repeated on the resuspended pellet. The OD of the resupended phage pellet was read at 270 nm.
[0480] The second, third and fourth rounds of phage sorting were completed by repeating the phage sorting steps as described above. The phage antibodies that were selected based on binding to the short form were designated as "S#" (e.g., S4, S9, S7 and S16). The phage antibodies that were selected based on binding to the full length form were designated as "F#" (e.g., F5, F6, F13 and F47).
[0481] ELISA Screening Assay: Clones from the sorts 2 to 4 were screened for specificity and affinity by ELISA assay. Positive clones (binders) were clones that had above background binding to the target antigens and not to other non-relevant protein, such as bovine serum albumin and insulin-like growth factor-1 (IGF-1).
[0482] First, the wells of a 384-well microtiter plate were coated with either full length or "short form" (#93aa-243aa) his-tagged human STOP-1, IGF-1, Her-2 or anti-gD at 20 ul per well (2 g/ml in PBS) at 4° C. overnight or room temperature for 2 hours.
TABLE-US-00022 HER2 STOP-1 Anti-gD IGF-1
In another 96 well plate, colonies from sorts 2-4 were grown overnight at 37° C. in 150 ul 50:50 2YT/CRAP media with 50 ug/ml carbenicillin and helper phage K07. The plate was spun down at 2500 rpm for 20 minutes. 50 ul of the supernatant was mixed with 120 ul of ELISA buffer (PBS--0.5% BSA and 0.05% Tween20). 30 ul of the mixture was added to each quadrant of the 384-well coating plate and incubated at room temperature for 1 hour. Binding was quantified by adding 75 ul/well of horse radish peroxidase (HRP)-conjugated anti-M13 antibody in PBS plus 0.5% BSA and 0.05% Tween20 at room temperature for 30 minutes (Sidhu et al., supra). The wells were washed with PBS--0.05% Tween20 at least five times. Next, 100 ul/well of a 1:1 ratio of 3,3',5,5'-tetramethylbenzidine (TMB) Peroxidase substrate and Peroxidase Solution B (H2O2) ((Kirkegaard-Perry Laboratories (Gaithersburg, Md.)) was added to the well and incubated for 5 minutes at room temperature. The reaction was stopped by adding 100 ul 1M Phosphoric Acid (H3PO4) ((Kirkegaard-Perry Laboratories (Gaithersburg, Md.)) to each well. The OD of the yellow color in each well was determined using a standard ELISA plate reader at 450 nm. The F(ab) or F(ab)'2 phage concentration that resulted in about 90% of maximum binding to the coated plate was used in the solution binding competition ELISAs. F(ab) and F(ab)'2 phage having 3-4 fold greater binding than BSA, IGF-1, Her2 and anti-gD were considered to have better specificity. Those binders were sequenced.
[0483] FIG. 18 shows a partial amino acid sequence of several of the binders that had higher affinity and specificity (e.g., S7, S16, F5, S4, F13, F47 and S9). Three clones share identical CDR sequences--1-F13, F47 and S4. S7 and S16 also share some sequence homology. Based on the sequence homology between F13, F47, S4, S7, S9 and S16 in their VH-CDR1, VH-CDR2 and VH-CDR3 regions, consensus sequences for the commonly recognized epitope were derived. Amino acid and nucleic acid sequences coding for a phage display S4-Fab, a phage display S9-Fab, a phage display S7-F(ab)'2, a phage display S16-F(ab)'2, a phage display F5-F(ab)'2 can be found in FIGS. 27A-C, FIGS. 28A-C, FIGS. 29A-C, FIGS. 30A-C and FIGS. 31A-C, respectively. S7 has SEQ ID NOS: 8-10. S16 has SEQ ID NOS:11-13. F5 has SEQ ID NOS: 14-16. S4, F13, and F47 have SEQ ID NOS: 17-19. S9 has SEQ ID NOS: 20-22.
[0484] The following vectors have been deposited under the terms of the Budapest Treaty with the American Type Culture Collection (ATCC), 10801 University Blvd., Manassas, Va. 20110-2209, USA as described below:
TABLE-US-00023 Material Deposit No. Deposit Date V0350-4-S7 PTA-5090 Mar. 25, 2003 V0350-4-S16 PTA-5089 Mar. 25, 2003 V0350-2b-S4 PTA-5086 Mar. 25, 2003 V0350-2b-S9 PTA-5087 Mar. 25, 2003 V0350-4-F5 PTA-5088 Mar. 25, 2003
Example 17
Solution Binding Competition ELISA
[0485] To determine a binding affinity for selected F(ab) and F(ab)'2 phage, competition ELISAs were performed.
[0486] First, the phage were propagated and purified. Ten uls of XL-1 bacteria infected with one of the clones for 30 minutes at 37° C. was plated on a carbenicillin plate. A colony was picked and grown in 2 mls (2YT and 50 ug/ml carbenicillin) at 37° C. for 3-4 hours. Helper phage, KO7, was added to the culture at a final concentration of 1010 pfu/ml for another 1 hour at 37° C. Twenty mls of media (2YT/CRAP 50:50 with 50 ug/ml carbenicillin was added to the culture for growth overnight at 37° C. The phage was purified as described above.
[0487] Second, the concentration of purified phage that would be optimal for use in the following competition ELISA assay was determined (i.e., approximately 90% of maximal binding capacity on the coated plate). 96-well Nunc Maxisorp plates were coated with full length or short form human STOP-1 (2 ug/ml in PBS) at 4° C. overnight or at room temperature for 2 hours. The wells were blocked by adding 65 ul 1% BSA for 30 minutes followed by 40 ul 1% Tween20 for another 30 minutes. Next, the wells were washed with PBS--0.05% Tween20 5 times. Various dilutions of F(ab) or F(ab)'2 phage down to 0.1 O.D./ml in ELISA buffer (PBS--0.1% BSA and 0.05% Tween20) were added to the wells for 15 minutes at room temperature. The wells were then washed with PBS--0.05% Tween20 at least three times. 75 ul of HRP-conjugated anti-M13 antibody (Amersham, 1/5000 dilution with ELISA buffer) per well was added and incubated at room temperature for 30 minutes. The wells were washed again with PBS--0.05% Tween20 at least five times. Next, 100 ul/well of a 1:1 ratio of 3,3',5,5'-tetramethylbenzidine (TMB) Peroxidase substrate and Peroxidase Solution B (H2O2) ((Kirkegaard-Perry Laboratories (Gaithersburg, Md.)) was added to the well and incubated for 5 minutes at room temperature. The reaction was stopped by adding 100 ul 1M Phosphoric Acid (H3PO4) ((Kirkegaard-Perry Laboratories (Gaithersburg, Md.)) to each well. The optical density of the color in each well was determined using a standard ELISA plate reader at 450 nm. The dilutions of phage were plotted against the O.D. readings.
[0488] Third, a competition ELISA was performed. 96-well Nunc Maxisorp plates were coated with full length or short form human STOP-1 (2 ug/ml in PBS) at 4° C. overnight or at room temperature for 2 hours. The wells were blocked by adding 65 ul 1% BSA for 30 minutes followed by 40 ul 1% Tween20 for another 30 minutes. The wells were washed with PBS--0.05% Tween20 5 times. Based on the binding assay above, 50 ul of the dilution of phage that resulted in about 90% of maximum binding to the coated plate was incubated with 50 ul of various concentrations of full length or short form human STOP-1 (0.1 to 500 nM) in ELISA buffer solution for 1 hour at room temperature in a well. The unbound phage was assayed by transferring 75 ul of the well mixture to second 96-well plate pre-coated with full length or short form human STOP-1 and incubating at room temperature for 15 minutes. The wells of the second plate were washed with PBS--0.5% Tween20 at least three times. 75 ul of HRP-conjugated anti-M13 antibody ( 1/5000 dilution with ELISA buffer) per well was added and incubated at room temperature for 30 minutes. The wells were washed again with PBS--0.05% Tween20 at least five times. Next, 100 ul/well of a 1:1 ratio of 3,3',5,5'-tetramethylbenzidine (TMB) Peroxidase substrate and Peroxidase Solution B (H2O2) ((Kirkegaard-Perry Laboratories (Gaithersburg, Md.)) was added to the well and incubated for 5 minutes at room temperature. The reaction was stopped by adding 100 ul 1M Phosphoric Acid (H3PO4) ((Kirkegaard-Perry Laboratories (Gaithersburg, Md.)) to each well. The optical density of the color in each well was determined using a standard ELISA plate reader at 450 nm. The concentrations of competitor STOP-1 were plotted against the O.D. readings. The IC50, the concentration of STOP-1 that inhibits 50% of the F(ab)-phage or F(ab)'2-phage, represents the affinity. See Table 12.
[0489] Table 12 shows that F5 binds the N-terminus of full length STOP-1, not the short form. S7, S16, S9 and S4 (and therefore F13 and F47) bind to the short form (#94-243) of human STOP-1. In Table 2, the term "762 S/S" indicates that the wells of the microtiter plate were coated with the short form of human STOP-1 and that the F(ab)-phage or F(ab)'2-phage were competed with the short form of human STOP-1. The term "762 S/F" indicates that the wells of the microtiter plate were coated with the short form of human STOP-1 and that the F(ab)-phage or F(ab)'2-phage were competed with the full length form of human STOP-1. The term "762 F/F" indicates that the wells of the microtiter plate were coated with the full length form of human STOP-1 and that the F(ab)-phage or F(ab)'2-phage were competed with the full length form of human STOP-1. "ND" indicates that the result was not detectable. "N/A" indicates that the result was not available.
TABLE-US-00024 TABLE 12 762 S/S 762 S/F 762 F/F STOP-1 binding F(ab)'2 S7 35 nM 4.7 nM 2.7 nM Short form S16 114 nM n/a 32 nM Short form F5 ND 1 uM N/A Full length Fab S4 3 nM 719 nM 0.9 nM Short form S9 >1 uM 331 nM N/A Short form F13 3.8 nM 795 nM n/a Short form F47 3.8 nM 795 nM n/a Short form
[0490] These antibodies also recognized human STOP-1 in immunoprecipitations.
Example 18
6B12 Blocking Assay
[0491] The binding location of certain F(ab)'2-phage was also explored. It is known that the monoclonal anti-human STOP-1 antibody, 6B12, binds to an N-terminal region of human STOP-1 protein. Therefore, it was tested whether 6B12 could block binding of certain F(ab)'2-phage to STOP-1.
[0492] The 6B12 blocking assay was conducted as follows: 96-well Nunc Maxisorp plates were coated with full length human STOP-1 (2 ug/ml in PBS) at 4° C. overnight or at room temperature for 2 hours. The wells were blocked by adding 65 ul 1% BSA at room temperature for 30 minutes followed by 40 ul 1% Tween20 for another 30 minutes. Next, the wells were washed with PBS--0.05% Tween20 5 times. Various concentrations of 6B12 antibody (in ELISA buffer) were incubated in the wells for 30 minutes at room temperature. Then, S7-F(ab)'2-phage, S16-F(ab)'2-phage or F5-F(ab)'2-phage were added to each well for 10 minutes at a concentration that would normally produce 90% binding capacity in the absence of the 6B12 antibody. The wells were washed with PBS--0.05% Tween20 5 times.
[0493] Binding was quantified by adding 75 ul/well of horse radish peroxidase (HRP)-conjugated anti-M13 antibody in PBS plus 0.5% BSA and 0.05% Tween20 at room temperature for 30 minutes (Sidhu et al., supra). The wells were washed with PBS--0.05% Tween20 at least five times. Next, 100 ul/well of a 1:1 ratio of 3,3',5,5'-tetramethylbenzidine (TMB) Peroxidase substrate and Peroxidase Solution B (H2O2) ((Kirkegaard-Perry Laboratories (Gaithersburg, Md.)) was added to the well. The reaction was stopped by adding 100 ul 1M Phosphoric Acid (H3PO4) ((Kirkegaard-Perry Laboratories (Gaithersburg, Md.)) to each well and allowed to incubate for 5 minutes at room temperature. The OD of the yellow color in each well was determined using a standard ELISA plate reader at 450 nm.
TABLE-US-00025 TABLE 13 F(ab)'2 6B12 blocking S7 - S16 - F5 +
[0494] Table 13 shows the 6B12 antibody was able to block F(ab)'2-F5 phage but not F(ab)'2-S7 phage or F(ab)'2-S16 phage. Therefore, F5 binds to human STOP-1 in the same N-terminal region as 6B12 whereas S7 and S16 do not.
Example 19
F(ab) and IgG Protein Constructs and Protein Expression
[0495] F(ab) Constructs for Expression in Bacterial Cells: V0350-2b-S4 and V0350-2b-S7 phagemids were modified by removing the viral cP3 sequences, replacing them with a terminator sequence containing 5'-GCTCGGTTGCCGCCGGGCGTTTTTTATG-3' (SEQ ID NO:85), and removing the sequences encoding the leucine zipper and gD tags (hereinafter, pv0120-54 and pV0120-57, respectively). FIG. 24C is a schematic of this vector. The pv0120 vectors were transformed into E. coli 34B8 cells. Single colonies were picked and grown in complete CRAP medium with 25 ug/ml carbenicillin at 30° C. for at least 22 hours. The expressed proteins were purified through a Protein G high trap column (Amersham Pharmacia).
[0496] Amino acid and nucleic acid sequences coding for an S4-Fab are illustrated in FIGS. 32A-G.
[0497] IgG Constructs for Expression in Mammalian Cells:
[0498] Generally, IgG1 constructs were made by swapping the light chain encoded in the LPG3 vector with the light chain of S4 or S7 and by swapping the VH and CH1 region encoded in the LPG4 vector with the VH and CH1 region of S4 or S7. FIG. 24D is a schematic of the LPG3 and LPG4 vectors encoding the light and heavy chains, respectively, of an IgG protein. The LPG3 vector encodes a humanized MaE11 E27 light chain. The LPG4 vector encodes a humanized MaE11 E27 heavy chain. Together, they encode a full-length human IgG1 version of humanized MaE11 E27 (an anti-IgE antibody). See, U.S. Pat. No. 6,172,213 (Lowman) for more information about humanized MaE11 E27. The LPG3 and LPG4 vectors were pRK vectors (Gorman, C M et al., (1990) DNA Protein Eng. Tech. 2:3) that were modified by, among other things, inserting the full-length light chain and heavy chain, respectively, of a humanized MaE11 E27. The LPG3 and LPG4 vectors were obtained from Yan Wu at Genentech, Inc., South San Francisco, Calif.
[0499] The LPG4 vector was digested with BsiwI and ApaI to remove the heavy chain variable regions of the humanized MaE11 E27 antibody. The removed sequences were replaced with a Bsiw1-ApaI fragment from the pv0120-54 or the pv0120-57 vectors. The LPG3 vector was digested with Ecor RV and KpnI to remove the light chain variable regions of the humanized MaE11 E27 antibody. The removed sequences were replaced with an Ecor RV-KpnI fragment from the pv0120-54 or the pv0120-57 vectors encoding the light chain variable regions of S4 and S7. The resulting vectors, LPG3-humankappaG6 and LPG4-humanHC-S4, are described in FIGS. 35 and 36 (SEQ ID NO:110 and SEQ ID NO:112, respectively). The sequence of LPG4-humanHC-S7 is the same as the sequence of LPG4-humanHC-S4, except that the sequence between the Bsiw1-ApaI sites is the same as the sequence between the Bsiw1-ApaI sites of the pv0120-57 vector.
[0500] LPG3-humankappaG6, LPG4-humanHC-S4 and LPG4-humanHC-S7 dsDNA were prepared for transfection. DP12 DHFR+ CHO cells (ATCC) were seeded at 1.5×106 cells/ml in tissue culture media containing 1× Tris EDTA (TE), 2 mg/L insulin, 1% dFBS, 0.15 g/L gentamycin sulfate. The cells were incubated at 37° C. for one to two hours before transfecting. Next, 3.5 L of warm tissue culture media was added to the culture together with 20 mg DNA, 20 ml of DMRIE-C reagent (1,2-dimyristyloxypropyl-3-dimethyl-hydroxyl ethyl ammonium bromide, Genentech, Inc.) and incubated for at least more 20 minutes at 37° C. The culture was added to a bioreactor, and 250 ml/L of warm tissue culture media was added. The cell culture temperature was shifted to 33° C. After 7-12 days, the cells were centrifuged at 1000 rpm for 5 minutes and then the supernatant was filtered through a 0.2 um filter. The proteins in the supernatant were purified through a Protein G high trap column (Amersham Pharmacia).
[0501] Amino acid and nucleic acid sequences coding for an S4 IgG protein are illustrated in FIGS. 33A-F and FIGS. 24A-G.
Example 20
Affinity Measurement of S4 and S7 Fab and IgG
[0502] ELISA assays were performed to determine the affinity of S4 and S7 Fab and IgG for human STOP-1. First, the concentration of purified Fab and IgG that would be optimal for use in a competition ELISA assay was determined (i.e., approximately 90% of maximal binding capacity on the coated plate). 96-well Nunc Maxisorp plates were coated with full length or short form human STOP-1 (2 ug/ml in PBS) at 4° C. overnight or at room temperature for 2 hours. The wells were blocked by adding 65 ul 1% BSA for 30 minutes followed by 40 ul 1% Tween20 for another 30 minutes. Next, the wells were washed with PBS--0.05% Tween20 5 times. Concentrations of F(ab) or IgG from 0.1 nM to 100 nM diluted in ELISA buffer (PBS--0.5% BSA and 0.05% Tween20) were added to the wells for 15 minutes at room temperature. The wells were then washed with PBS--0.05% Tween20 at least three times. 75 ul of HRP-conjugated Protein G antibody (Amersham, 1/5000 dilution with ELISA buffer) per well was added and incubated at room temperature for 30 minutes. The wells were washed again with PBS--0.05% Tween20 at least five times. Next, 100 ul/well of a 1:1 ratio of 3,3',5,5'-tetramethylbenzidine (TMB) Peroxidase substrate and Peroxidase Solution B (H2O2) ((Kirkegaard-Perry Laboratories (Gaithersburg, Md.)) was added to the well. The reaction was stopped by adding 100 ul 1M Phosphoric Acid (H3PO4) ((Kirkegaard-Perry Laboratories (Gaithersburg, Md.)) to each well and allowed to incubate for 5 minutes at room temperature. The optical density of the color in each well was determined using a standard ELISA plate reader at 450 nm. The dilutions of Fab or IgG were plotted against the O.D. readings.
[0503] FIG. 19 shows an example of a ELISA assay used to determine the optimal concentration of Fab or IgG in a competition assay. "S coated" refers to a short form (#94-243) of STOP-1 coated on a microtiter plate. "F coated" refers to a full-length form of human STOP-1 coated on a microtiter plate. Approximately 90% of maximal binding was considered to be optimal for use in a competitive ELISA assay.
[0504] Next, a competition ELISA was performed using the optimal concentration of Fab and IgG determined above. 96-well Nunc Maxisorp plates were coated with full length or short form human STOP-1 (2 ug/ml in PBS) at 4° C. overnight or at room temperature for 2 hours. The wells were blocked by adding 65 ul 1% BSA for 30 minutes followed by 40 ul 1% Tween20 for another 30 minutes. The wells were washed with PBS--0.05% Tween20 5 times. Based on the binding assay above, 50 ul of the dilution of Fab or IgG that resulted in about 90% of maximum binding to the coated plate was incubated with 50 ul of various concentrations of full length or short form human STOP-1 (0.1 to 500 nM) in ELISA buffer solution for 1 hour at room temperature in a well. The unbound Fab or IgG was assayed by transferring 75 ul of the well mixture to second 96-well plate pre-coated with full length or short form human STOP-1 and incubating at room temperature for 15 minutes. The wells of the second plate were washed with PBS--0.5% Tween20 at least three times. 75 ul of HRP-conjugated Protein G ( 1/5000 dilution with ELISA buffer) per well was added and incubated at room temperature for 30 minutes. The wells were washed again with PBS--0.05% Tween20 at least five times. Next, 100 ul/well of a 1:1 ratio of 3,3',5,5'-tetramethylbenzidine (TMB) Peroxidase substrate and Peroxidase Solution B (H2O2) ((Kirkegaard-Perry Laboratories (Gaithersburg, Md.)) was added to the well. The reaction was stopped by adding 100 ul 1M Phosphoric Acid (H3PO4) ((Kirkegaard-Perry Laboratories (Gaithersburg, Md.)) to each well and allowed to incubate for 5 minutes at room temperature. The optical density of the color in each well was determined using a standard ELISA plate reader at 450 nm. The concentrations of the added competitor STOP-1 were plotted against the O.D. readings. The IC50 is the concentration of STOP-1 that inhibited 50% of the Fab or IgG binding. See FIGS. 20A and B. The binding affinities are indicated in the parentheticals.
[0505] FIG. 21 is a summary of the binding affinities of several phage-derived antibodies against STOP-1. "S/S" refers to an ELISA in which the microtiter plate was coated with a short form of STOP-1 and competed with a short form of STOP-1. "F/S" refers to an ELISA in which the microtiter plate was coated with a full-length form of human STOP-1 and competed with a short form of human STOP-1. "F/F" refers to an ELISA in which the microtiter plate was coated with a full-length form of STOP-1 and competed with a full-length form of STOP-1. The phage used in these studies were the S4-Fab phage and the S7-F(ab)'2 phage.
Example 21
S4 Blocking Assay
[0506] S4 IgG was used in a competitive ELISA assay to see if it could block S4 (Fab)-phage, S7 (F(ab)'2)-phage, S9 (Fab)-phage, S16 (F(ab)'2)-phage or F5 (F(ab)'2)-phage binding to a short form or a full-length form of human STOP-1.
[0507] First, 96-well Nunc Maxisorp plates were coated with full length human STOP-1 (2 ug/ml in PBS) at 4° C. overnight or at room temperature for 2 hours. The wells were blocked by adding 65 ul 1% BSA for 30 minutes followed by 40 ul 1% Tween20 for another 30 minutes. Next, the wells were washed with PBS--0.05% Tween20 5 times. Various concentrations of S4 IgG (in ELISA buffer) were incubated in the wells for 30 minutes at room temperature. Then, S4 (Fab)-phage, S7 (F(ab)'2)-phage, S9 (Fab)-phage, S16 (F(ab)'2)-phage or F5 (F(ab)'2)-phage were added to different wells for 10 minutes at a concentration that would normally produce 90% binding capacity in the absence of the S4 IgG antibody. The wells were washed with PBS--0.05% Tween20 5 times.
[0508] Binding was quantified by adding 75 ul/well of horse radish peroxidase (HRP)-conjugated anti-M13 antibody in PBS plus 0.5% BSA and 0.05% Tween20 at room temperature for 30 minutes (Sidhu et al., supra). The wells were washed with PBS--0.05% Tween20 at least five times. Next, 100 ul/well of a 1:1 ratio of 3,3',5,5'-tetramethylbenzidine (TMB) Peroxidase substrate and Peroxidase Solution B (H2O2) ((Kirkegaard-Perry Laboratories (Gaithersburg, Md.)) was added to the well. The reaction was stopped by adding 100 ul 1M Phosphoric Acid (H3PO4) ((Kirkegaard-Perry Laboratories (Gaithersburg, Md.)) to each well. The OD of the yellow color in each well was determined using a standard ELISA plate reader at 450 nm.
[0509] FIG. 22 shows that most of the phage-derived antibodies, except F5, bound a similar region on STOP-1 as the S4 phage-derived antibody. The Y axis refers to percentage unblocked as calculated by dividing the OD450 nm value of the well that blocked S4 IgG by the OD450 nm value of a well without S4 IgG. The phage tested were S4 (Fab) phage, S7 (F(ab)'2) phage, S9 (Fab) phage, S16 (F(ab)'2) phage and F5 (F(ab)'2) phage. A lower % means the more blocking by S4 IgG.
Example 23
Optimizing Binding By Altering The Light Chain Sequence
[0510] Binding of the antibodies can be further optimized by, inter alia, by altering the sequence of the light chain. Optimization can be carried out by methods known in the art, including known phage display methods. Additionally, the sequence of the light chain CDR's can be changed by site-directed mutagenesis and screened by ELISA assays similar to the methods described in Example 16 above, except that the diversity is generated in the light chain variable region as described in U.S. Provisional Patent No. 60/385,388, filed Jun. 3, 2002. See also below.
[0511] According to one example, the libraries of antibody variable light chain domains are optimized to maximize diversity in the CDR regions while minimizing structural perturbations in the antibody variable domains Structural perturbations in antibody variable domains are generally associated with improperly folded antibody domains resulting in low yield, for example when produced in bacterial cells. Low yields decrease the number of binders detected in screening. Diversity in the light chain CDR regions can be generated by identifying solvent accessible and highly diverse positions in each CDR for CDRs L1, L2, L3, and designing an oligonucleotide comprising at least one tailored (i.e., non-random) codon set encoding variant amino acids for the amino acid position corresponding to the position of at least one solvent accessible residue at a highly diverse position in at least one CDR region. A tailored codon set is a degenerate nucleic acid sequence that preferably encodes the most commonly occurring amino acids at the corresponding positions of the solvent accessible residues in known, natural antibodies.
[0512] Solvent accessible residues in the CDRs can be identified in the antibody variable domain template molecule by analyzing the crystal structure of the template molecule. Humanized antibody 4D5 is efficiently produced and properly folded when produced in a variety of host cells, including bacterial cell culture. The crystal structure for the humanized antibody 4D5 variable region is known and publicly available at http://www.rcsb.org (accession code IFVC).
[0513] The solvent accessible positions in the CDRs of the light chain have been identified using the Insight II program (Accelrys, San Diego, Calif.).
[0514] CDR residues were also analyzed to determine which positions in the CDRs were highly diverse. Highly diverse positions in the CDR regions for the heavy and light chains were identified by examining the sequences of known, naturally occurring antibodies in the Kabat database (Kabat, E. A., et al., (1991) Sequences of Proteins of Immunological Interest, Fifth Edition, NIH Publication No. 91-3242). The Kabat database is also available through http://www.bioinf.org.uk/abs/. In the Kabat database, there were about 1540 sequences of the human light chain and 3600 sequences for the human heavy chain. The CDR sites were aligned and numbered as described by Kabat (see http://www.bioinf.org.uk/abs/du). Highly diverse amino acid positions were identified by lining up and ranking the amino acid usage, from most frequently used to less frequently used for each CDR residue. For example, L3-91 (i.e., residue 91 of the light chain CDR3) was found to be Y (tyrosine) in 849 out of 1582 antibody sequences in the Kabat database, and it is the amino acid found most frequently at this position. Next on the list of frequency serine (occurring in 196 sequences), followed by arginine (169 sequences), alanine (118 sequences), glycine (61 sequences), histidine (41 sequences), with the remaining 35 sequences being one of the remaining amino acids. The frequency of amino acids in human antibody light chain sequences from the Kabat database (including illustrative diverse sites, with corresponding diversity list of amino acids) is shown in FIG. 35.
[0515] Amino acid residues found in a particular position that collectively constitute the most frequently occurring amino acids among the known, natural antibody sequences can be selected as the basis for library design. The most frequently occurring amino acids were deemed to be those that most commonly found in the top 90% of the list of diverse amino acids (this group of amino acids is referred to herein as "target group of amino acids"). However, as described herein, the percent cutoff for a target group of amino acids can be varied, as described above, according to the circumstances and purpose of the diversity library that is to be achieved.
[0516] For humanized antibody 4D5, the positions identified as solvent accessible and highly diverse were:
TABLE-US-00026 Light Chain CDR1 28, 29, 30, 31, 32 CDR2 50, 53 CDR3 91, 92, 93, 94, 96 Heavy Chain CDR 1 28, 30, 31, 32, 33 CDR2 50, 52, 53, 54, 56, 58
[0517] Examples of amino acids that occur at high frequency in natural diversity (i.e., among known, natural antibody sequences) (referred to as "target group" or "natural diversity" in FIG. 3), and the designed diversity of amino acids by DNA codons ("Diversity <DNA codon>") for each of these positions is shown in FIG. 36.
[0518] Codon sets encoding a specific group of amino acids (Diversity) have been designed to include at least a certain percentage of the amino acids in the known, natural sequences (designated as "% covering" in FIG. 36). Of the amino acids encoded by a codon set, at least about 40% of the amino acid can target amino acids identified for a particular solvent accessible and highly diverse position (designated as "% good" in FIG. 36; amino acids encoded by a codon set that are target amino acids are shown in bold in column 3 of FIG. 36). However, as described herein, the % good value can be varied according to circumstance and objectives. The codon sets were selected such that they preferably encoded the amino acids with the highest occurrences at a particular position. The number of non-target amino acids coded by a codon set for a particular position was minimized. Effectiveness of codon set selection/design was evaluated in part based on the "% good" value. A high percentage meant very low non-target amino acids; a high value of "% good" was deemed more important than having more target amino acids among the amino acids coded by a particular codon set. Redundancy was included in calculating the "% good" value. For evaluation purposes, the "% covering" value was also calculated. This value represents the percentage of natural diversity covered by the "good" amino acids (of the amino acids encoded by a particular codon set). For example, for L3-91, when codon set KMT is used, the "good" amino acids are YSA, which is 75% of the YSAD amino acids encoded by the codon. YSA are amino acids that cover 1190 out of 1580 known, natural antibody sequences at this amino acid position. 1190/1580 equals 75%, which is the "% covering" value. Thus, in one design using KMT at L3-91, 75% of the library covers 75% of the natural diversity in CDRL3 at position 91.
[0519] The codon sets were also designed to exclude, when possible, cysteine and stop codons. The presence of cysteine residues tends to cause folding problems and stop codons can decrease the effective library size. In the design of the codon sets, it was also deemed desirable to minimize the number of nontarget amino acids.
[0520] The codon sets designed for each solvent accessible and highly diverse residue of humanized antibody 4D5 are shown in FIG. 36. At any particular residue, one or more codon set(s) could be used depending on the target amino acids that are identified. For example, two L1 oligonucleotides can be combined--one containing codon YKG and the other containing TWT at L3-96, or one containing codon DGG and the other containing DHT at H2-50.
[0521] The various codon sets could be used to generate diverse libraries with diversity in one or more CDR regions, including CDR L1, CDR L2, CDR L3. For example, FIGS. 37-40 show various illustrative versions of codon set designs that can be used to generate diversity. FIG. 36 provides a summary of the amino acid coverage of these designs. In general, it is preferable, but not necessary, that the designs narrow the diversity to cover more of the natural diversity and exclude as much as possible the "non-target" amino acids. In some embodiments, a design that does not score the highest based on these criteria can be used to obtain good binders for a STOP-1.
Example 24
STOP-1 Binds to the Cell Surface of Cells
[0522] HT1080 cells were incubated with recombinant purified full length human His-tagged STOP-1 (10 ug/ml, A; 0.5 million cells per sample) in the presence of 100 ug/ml of S7 or 6b12 monoclonal antibodies in FACS buffer (20 mM HEPES, pH 7.5, 140 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, 2% FBS) at 4° C. for 1 hour. Protein binding was detected by treating the cells with FACS with anti-His monoclonal antibodies (5 mg/ml) or anti-Flag (5 mg/ml, as a negative control) and followed by FITC-conjugated goat anti-mouse antibodies.
[0523] FIG. 41 shows that STOP-1 binds specifically to the surface of human HeLa cells, human HT1080 fibroblast cells and human umbilical vein endothelial cells (HUVEC) cells, but not human embryonic kidney 293 cells. It is believed that a receptor for STOP-1 exists on HeLa, HT1080 and HUVEC cells.
Example 25
S7 Monoclonal Antibody Promotes STOP-1 Binding to Cells
[0524] HT1080 cells were incubated with recombinant purified full length human His-tagged STOP-1 (10 ug/ml, A; 0.5 million cells per sample) in presence of 100 ug/ml of S7 or 6b12 monoclonal antibodies in FACS buffer (20 mM HEPES, pH 7.5, 140 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, 2% FBS) at 4° C. for 1 hour. Protein binding was detected by FACS treatment with anti-His monoclonal antibodies (5 mg/ml) or anti-Flag (5 mg/ml, as a control) followed by treatment with FITC-conjugated goat anti-mouse antibodies.
[0525] FIG. 42 shows that the S7 antibody potentiated STOP-1 binding to the cell surface of HT1080 cells whereas 6B12 did not. The S4 antibody was also tested in the same assay and found to potentiate STOP-1 binding to HT1080 cells. Because the S7 antibodies and S4 antibodies were able to bind to STOP-1 bound to cells, the binding of S7 antibody or S4 antibody to STOP-1 does not appear to interfere with the binding of the STOP-1 receptor to STOP-1. Thus, the epitope that the S7 antibody binds to on STOP-1 does not appear to be required for STOP-1 binding to its receptor.
Example 26
STOP-1 Promotes Endothelial Cell Migration
[0526] Directional cell migration was measured using a modified Boyden chemotaxis chamber (Transwells®, Corning, Inc.). Polycarbonate filters with 8 micron pores were incubated with a 0.1% solution of collagen I from Collaborative Sciences overnight at 4° C. This process coated the undersurface of the filters with collagen, an extracellular matrix protein necessary for cell attachment. The next day, the filters were rinsed in PBS and blocked for 1 hr at room temperature with a blocking medium (200 microliters in each lower chamber) that consisted of basic endothelial cell medium (Clonetics) with 1% bovine serum albumin (BSA).
[0527] HUVEC cells (from Clonetics, grown in complete medium) were harvested using a cell dissociation solution (0.25% EDTA) and resuspended in a migration medium (endothelial cell basic medium having 0.1% BSA, but no growth factors). The resuspended cells were placed in the upper chamber of the modified Boyden chamber (10,000 cells/ml, 150 microliters/well). The migration medium containing bFGF (1 ng/ml or 10 ng/ml, final concentration) or STOP-1 (1 ug/ml or 10 ug/ml, final concentration) were added to the lower chamber of the modified Boyden chamber (300 microliters/well). Migration medium alone (without bFGF or STOP-1) was added to the lower chambers as controls. Each condition was done in triplicate. The modified Boyden chemotaxis chambers were placed in an incubator at 37° C. (5% CO2) for 3 hours.
[0528] Afterwards, cells that had not migrated (located on the top of the filters facing the upper chamber) were gently removed using cotton swabs. The filters were fixed in a 4% paraformaldehyde solution (10 min, RT) and stained with a 0.2% crystal violet solution (5 min, RT), to visualize the cells. The cells located on the undersurface of the filters (i.e., that migrated) were then counted using a Nikon inverted microscope. The filters were randomized ("blinding" the investigator to the conditions). The cells in six random fields (40×) per filter were counted, 3 filters per condition. The data in FIG. 44 represents the mean number of cells from one representative experiment (out of 3).
[0529] FIG. 43 indicates that STOP-1 is chemotactic (i.e., induces directional migration) to HUVEC cells. This effect is comparable in magnitude to that of bFGF (a growth factor which has been previously shown to induce HUVEC migration and to act overall as a pro-angiogenic molecule). Treatment with both bFGF and STOP-1 did not show an additive effect. Nor did treatment with bFGF or STOP-1 potentiate each other's effects. This preliminary data suggests that STOP-1 acts as a pro-angiogenenic or pro-vasculogenic molecule. Work to further confirm this effect will include the repetition of the same or similar experiment using an antibody that blocks of the interaction of STOP-1 with HUVEC cells.
Example 27
STOP-1 Binds to MDA435 Cells
[0530] MDA-MB-435 human mammary carcinoma cells were removed from culture flasks with 10 mM EDTA and resuspended in cold PBS containing 2% fetal bovine serum. The cell number was adjusted to 1,000,000 cells/ml, and 0.5 ml of the suspension was dispensed into tubes with 10 mg/ml flag-tagged full length human STOP-1 and 100 mg/ml of specific (6B12) or control (4B7) antibodies. The mixture was incubated for 1 hour on ice and washed with cold suspension buffer. The mixture was then incubated with anti-flag M2-FITC for 1 hour on ice. Cells were washed in suspension buffer and fluorescence was measured by flow cytometry.
[0531] FIG. 44 graphically depicts the flow cytometry analysis. The geometric mean value of the signal intensity produced by cells treated with detection antibody alone (anti-flag M2-FITC antibody without STOP-1) was approximately 7.99. The geometric mean values of the signal intensities produced by the cells treated with STOP-1 alone, STOP-1 with 6B12 and STOP-1 with 4B7 were approximately 13.24, 8.3 and 11.39, respectively. The results show that STOP-1 binds to MDA-MB-435 breast carcinoma cells and that 6B12, a monoclonal antibody specific to STOP-1, blocks STOP-1 binding to those cells. The isotype control, 4B7 antibody, showed no appreciable activity. S4 and S7 antibodies were also used in this assay. Both antibodies did not block STOP-1 from binding to MDA-MB-435 cells, their presence in the assay resulted in an increase in the STOP-1 binding to the cells.
Example 28
STOP-1 mRNA Expression is Upregulated by TNFalpha and Cellular Stress
[0532] Previous studies have shown that conditions of low oxygen (hypoxia) or cellular stress promote tumorigenesis and angiogenesis. TNFalpha is often used to stimulate cellular responses associated with stress and has been implicated in promoting angiogenesis and tumorigenesis. To test whether STOP-1 expression was influenced by tumorigenic and angiogenic triggers, HUVEC cells were subjected to treatment with TNFalpha and hypoxic conditions.
[0533] HUVEC cells were incubated for 3, 8 or 24 hours with 100 ng/ml of human recombinant TNFalpha (Genentech, Inc.) under hypoxic conditions (95% air, 5% CO2) or normoxic conditions (approximately, 95% air, 5% CO2). The mRNA from the treated cells was extracted and subjected to TaqMan RT-PCR reactions using the primers and conditions described in Example 2. The fold change in expression of STOP-1 mRNA at the end of each time period was calculated by comparing it to its level of expression after three hours of incubation under normoxic conditions (without 100 ng/ml TNFalpha).
[0534] FIG. 45B shows that STOP-1 mRNA expression is significantly upregulated upon treatment with TNFalpha under normoxic conditions. The expression levels did not appear to change substantially at the 3, 8 and 34 hour time points. FIG. 45A, on the other hand, shows that TNFalpha has little or no effect on STOP-1 mRNA levels in cells incubated under hypoxic conditions. Interestingly, in absence of TNFalpha, STOP-1 expression was significantly upregulated after 34 hours of treatment under hypoxic conditions, but not after 3 hours of treatment. These results indicate that STOP-1 expression in HUVEC cells is responsive to treatment with TNFalpha and hypoxic conditions, but the response is not additive or synergistic, possibly indicating similar pathways of action.
Sequence Listing Key
TABLE-US-00027 [0535] SEQ ID Description 1 DNA76393-1664 2 amino acid sequence of DNA76393-1664 3 alternative STOP-1 amino acid sequence 4 mouse STOP-1 amino acid sequence 5 rice fish STOP-1 amino acid sequence 6 zebra fish STOP-1 amino acid sequence 7 chicken STOP-1 amino acid sequence 8 S7 - first amino acid sequence 9 S7 - second amino acid sequence 10 S7 - third amino acid sequence 11 S16 - first amino acid sequence 12 S16 - second amino acid sequence 13 S16 - third amino acid sequence 14 F5 - first amino acid sequence 15 F5 - second amino acid sequence 16 F5 - third amino acid sequence 17 S4 - first amino acid sequence 18 S4 - second amino acid sequence 19 S4 - third amino acid sequence 20 S9 - first amino acid sequence 21 S9 - second amino acid sequence 22 S9 - third amino acid sequence 23 RT-PCR hybridization probe 24 RT-PCR forward primer 25 RT-PCR reverse primer 26 Template sequence 27 Primer sequence 28 Primer sequence 29 Primer sequence 30 Primer sequence 31 Primer sequence 32 Primer sequence 33 Primer sequence 34 Primer sequence 35 Primer sequence 36 Primer sequence 37 Primer sequence 38 Primer sequence 39 Primer sequence 40 Primer sequence 41 Primer sequence 42 Primer sequence 43 Primer sequence 44 Primer sequence 45 Primer sequence 46 Primer sequence 47 Primer sequence 48 Primer sequence 49 Primer sequence 50 Primer sequence 51 Primer sequence 52 Primer sequence 53 Primer sequence 54 Primer sequence 55 Primer sequence 56 Primer sequence 57 Primer sequence 58 Primer sequence 59 Primer sequence 60 Primer sequence 61 Primer sequence 62 Primer sequence 63 Primer sequence 64 Primer sequence 65 Primer sequence 66 Primer sequence 67 Primer sequence 68 Primer sequence 69 Primer sequence 70 Primer sequence 71 Primer sequence 72 Primer sequence 73 Primer sequence 74 Primer sequence 75 Primer sequence 76 Primer sequence 77 Primer sequence 78 Primer sequence 79 Primer sequence 80 Primer sequence 81 Primer sequence 82 Primer sequence 83 Primer sequence 84 GCN4 leucine zipper 85 Oligo containing terminator sequence 86 an amino acid sequence comprising an anti-Her-2 Fab light chain 87 an amino acid sequence comprising an anti-Her-2 Fab heavy chain region 88 a nucleic acid sequence of a phagemid encoding SEQ ID NOs: 86 and 87 (pv0350-2b) 89 an amino acid sequence comprising an anti-Her-2 F(ab)'2 light chain 90 an amino acid sequence comprising an anti-Her-2 F(ab)'2 heavy chain region 91 a nucleic acid sequence of a phagemid encoding the amino acid sequences of SEQ ID NOs: 89 and 90 (pv0350-4) 92 an amino acid sequence comprising an S4-Fab light chain 93 an amino acid sequence comprising an S4-Fab heavy chain region 94 a nucleic acid sequence encoding the amino acid sequences of SEQ ID NOs: 92 and 93 95 an amino acid sequence comprising an S9-Fab light chain 96 an amino acid sequence comprising an S9-Fab heavy chain region 97 a nucleic acid sequence encoding the amino acid sequences of SEQ ID NOs: 95 and 96 98 an amino acid sequence comprising an S7-F(ab)'2 light chain 99 an amino acid sequence comprising an S7-F(ab)'2 heavy chain region 100 a nucleic acid sequence encoding the amino acid sequences of SEQ ID NOs: 98 and 99 101 an amino acid sequence comprising an S16-F(ab)'2 light chain 102 an amino acid sequence comprising an S16-F(ab)'2 heavy chain region 103 a nucleic acid sequence encoding the amino acid sequences of SEQ ID NOs: 101 and 102 104 an amino acid sequence comprising a F5-F(ab)'2 light chain 105 an amino acid sequence comprising an F5-F(ab)'2 heavy chain region 106 a nucleic acid sequence encoding the amino acid sequences of SEQ ID NOs: 104 and 105 107 an amino acid sequence comprising an S4-Fab light chain 108 an amino acid sequence comprising an S4-Fab heavy chain region 109 a nucleic acid sequence of a vector encoding the amino acid sequence if SEQ ID NOs: 107 and 108 (pv0120-S4) 110 an amino acid sequence comprising an S4 IgG Light Chain 111 a nucleic acid sequence of a vector encoding the amino acid sequence of SEQ ID NO: 110 (LPG3.HumanKappaG6) 112 an amino acid sequence comprising an S4 IgG Heavy Chain 113 nucleic acid sequence of a vector encoding the amino acid sequence of SEQ ID NO: 112 (LPG4.HumanHC-S4) 114 Consensus Amino Acid Sequence of FIG. 1 115 An H2 consensus sequence 116 An H3 consensus sequence 117 Polypeptide sequence
All patent, applications and publications recited herein are hereby incorporated by reference.
Sequence CWU
1
11711257DNAHomo sapiens 1ggagagaggc gcgcgggtga aaggcgcatt gatgcagcct
gcggcggcct cggagcgcgg 60cggagccaga cgctgaccac gttcctctcc tcggtctcct
ccgcctccag ctccgcgctg 120cccggcagcc gggagccatg cgaccccagg gccccgccgc
ctccccgcag cggctccgcg 180gcctcctgct gctcctgctg ctgcagctgc ccgcgccgtc
gagcgcctct gagatcccca 240aggggaagca aaaggcgcag ctccggcaga gggaggtggt
ggacctgtat aatggaatgt 300gcttacaagg gccagcagga gtgcctggtc gagacgggag
ccctggggcc aatgttattc 360cgggtacacc tgggatccca ggtcgggatg gattcaaagg
agaaaagggg gaatgtctga 420gggaaagctt tgaggagtcc tggacaccca actacaagca
gtgttcatgg agttcattga 480attatggcat agatcttggg aaaattgcgg agtgtacatt
tacaaagatg cgttcaaata 540gtgctctaag agttttgttc agtggctcac ttcggctaaa
atgcagaaat gcatgctgtc 600agcgttggta tttcacattc aatggagctg aatgttcagg
acctcttccc attgaagcta 660taatttattt ggaccaagga agccctgaaa tgaattcaac
aattaatatt catcgcactt 720cttctgtgga aggactttgt gaaggaattg gtgctggatt
agtggatgtt gctatctggg 780ttggcacttg ttcagattac ccaaaaggag atgcttctac
tggatggaat tcagtttctc 840gcatcattat tgaagaacta ccaaaataaa tgctttaatt
ttcatttgct acctcttttt 900ttattatgcc ttggaatggt tcacttaaat gacattttaa
ataagtttat gtatacatct 960gaatgaaaag caaagctaaa tatgtttaca gaccaaagtg
tgatttcaca ctgtttttaa 1020atctagcatt attcattttg cttcaatcaa aagtggtttc
aatatttttt ttagttggtt 1080agaatacttt cttcatagtc acattctctc aacctataat
ttggaatatt gttgtggtct 1140tttgtttttt ctcttagtat agcattttta aaaaaatata
aaagctacca atctttgtac 1200aatttgtaaa tgttaagaat tttttttata tctgttaaat
aaaaattatt tccaaca 12572243PRTHomo sapiens 2Met Arg Pro Gln Gly Pro
Ala Ala Ser Pro Gln Arg Leu Arg Gly Leu1 5
10 15Leu Leu Leu Leu Leu Leu Gln Leu Pro Ala Pro Ser
Ser Ala Ser Glu 20 25 30Ile
Pro Lys Gly Lys Gln Lys Ala Gln Leu Arg Gln Arg Glu Val Val 35
40 45Asp Leu Tyr Asn Gly Met Cys Leu Gln
Gly Pro Ala Gly Val Pro Gly 50 55
60Arg Asp Gly Ser Pro Gly Ala Asn Val Ile Pro Gly Thr Pro Gly Ile65
70 75 80Pro Gly Arg Asp Gly
Phe Lys Gly Glu Lys Gly Glu Cys Leu Arg Glu 85
90 95Ser Phe Glu Glu Ser Trp Thr Pro Asn Tyr Lys
Gln Cys Ser Trp Ser 100 105
110Ser Leu Asn Tyr Gly Ile Asp Leu Gly Lys Ile Ala Glu Cys Thr Phe
115 120 125Thr Lys Met Arg Ser Asn Ser
Ala Leu Arg Val Leu Phe Ser Gly Ser 130 135
140Leu Arg Leu Lys Cys Arg Asn Ala Cys Cys Gln Arg Trp Tyr Phe
Thr145 150 155 160Phe Asn
Gly Ala Glu Cys Ser Gly Pro Leu Pro Ile Glu Ala Ile Ile
165 170 175Tyr Leu Asp Gln Gly Ser Pro
Glu Met Asn Ser Thr Ile Asn Ile His 180 185
190Arg Thr Ser Ser Val Glu Gly Leu Cys Glu Gly Ile Gly Ala
Gly Leu 195 200 205Val Asp Val Ala
Ile Trp Val Gly Thr Cys Ser Asp Tyr Pro Lys Gly 210
215 220Asp Ala Ser Thr Gly Trp Asn Ser Val Ser Arg Ile
Ile Ile Glu Glu225 230 235
240Leu Pro Lys3243PRTHomo sapiens 3Met Arg Pro Gln Gly Pro Ala Ala Ser
Pro Gln Arg Leu Arg Gly Leu1 5 10
15Leu Leu Leu Leu Leu Leu Gln Leu Pro Ala Pro Ser Ser Ala Ser
Glu 20 25 30Ile Pro Lys Gly
Lys Gln Lys Ala Gln Leu Arg Gln Arg Glu Val Val 35
40 45Asp Leu Tyr Asn Gly Met Cys Leu Gln Gly Pro Ala
Gly Val Pro Gly 50 55 60Arg Asp Gly
Ser Pro Gly Ala Asn Gly Ile Pro Gly Thr Pro Gly Ile65 70
75 80Pro Gly Arg Asp Gly Phe Lys Gly
Glu Lys Gly Glu Cys Leu Arg Glu 85 90
95Ser Phe Glu Glu Ser Trp Thr Pro Asn Tyr Lys Gln Cys Ser
Trp Ser 100 105 110Ser Leu Asn
Tyr Gly Ile Asp Leu Gly Lys Ile Ala Glu Cys Thr Phe 115
120 125Thr Lys Met Arg Ser Asn Ser Ala Leu Arg Val
Leu Phe Ser Gly Ser 130 135 140Leu Arg
Leu Lys Cys Arg Asn Ala Cys Cys Gln Arg Trp Tyr Phe Thr145
150 155 160Phe Asn Gly Ala Glu Cys Ser
Gly Pro Leu Pro Ile Glu Ala Ile Ile 165
170 175Tyr Leu Asp Gln Gly Ser Pro Glu Met Asn Ser Thr
Ile Asn Ile His 180 185 190Arg
Thr Ser Ser Val Glu Gly Leu Cys Glu Gly Ile Gly Ala Gly Leu 195
200 205Val Asp Val Ala Ile Trp Val Gly Thr
Cys Ser Asp Tyr Pro Lys Gly 210 215
220Asp Ala Ser Thr Gly Trp Asn Ser Val Ser Arg Ile Ile Ile Glu Glu225
230 235 240Leu Pro
Lys4245PRTHomo sapiens 4Met His Pro Gln Gly Arg Ala Ala Pro Pro Gln Leu
Leu Leu Gly Leu1 5 10
15Phe Leu Val Leu Leu Leu Leu Leu Gln Leu Ser Ala Pro Ser Ser Ala
20 25 30Ser Glu Asn Pro Lys Val Lys
Gln Lys Ala Leu Ile Arg Gln Arg Glu 35 40
45Val Val Asp Leu Tyr Asn Gly Met Cys Leu Gln Gly Pro Ala Gly
Val 50 55 60Pro Gly Arg Asp Gly Ser
Pro Gly Ala Asn Gly Ile Pro Gly Thr Pro65 70
75 80Gly Ile Pro Gly Arg Asp Gly Phe Lys Gly Glu
Lys Gly Glu Cys Leu 85 90
95Arg Glu Ser Phe Glu Glu Ser Trp Thr Pro Asn Tyr Lys Gln Cys Ser
100 105 110Trp Ser Ser Leu Asn Tyr
Gly Ile Asp Leu Gly Lys Ile Ala Glu Cys 115 120
125Thr Phe Thr Lys Met Arg Ser Asn Ser Ala Leu Arg Val Leu
Phe Ser 130 135 140Gly Ser Leu Arg Leu
Lys Cys Arg Asn Ala Cys Cys Gln Arg Trp Tyr145 150
155 160Phe Thr Phe Asn Gly Ala Glu Cys Ser Gly
Pro Leu Pro Ile Glu Ala 165 170
175Ile Ile Tyr Leu Asp Gln Gly Ser Pro Glu Leu Asn Ser Thr Ile Asn
180 185 190Ile His Arg Thr Ser
Ser Val Glu Gly Leu Cys Glu Gly Ile Gly Ala 195
200 205Gly Leu Val Asp Val Ala Ile Trp Val Gly Thr Cys
Ser Asp Tyr Pro 210 215 220Lys Gly Asp
Ala Ser Thr Gly Trp Asn Ser Val Ser Arg Ile Ile Ile225
230 235 240Glu Glu Leu Pro Lys
2455229PRTOryzias latipes 5Met Thr Pro Leu Ser Pro Arg Leu Leu Ile
Leu Leu Cys Leu Ala Leu1 5 10
15Pro Leu His Gly Gln Glu Lys Gly Arg Ser Arg Gly Tyr Arg Lys Asp
20 25 30Pro Asp Ala Asp Lys Phe
Gly Ser Cys Leu Gln Gly Pro Ala Gly Thr 35 40
45Pro Gly Arg Asp Gly Asn Pro Gly Ala Asn Gly Ile Pro Gly
Thr Pro 50 55 60Gly Ile Pro Gly Arg
Asp Gly Leu Lys Gly Glu Lys Gly Glu Cys Val65 70
75 80Ser Glu Val Phe Glu Glu Pro Trp Lys Pro
Asn Tyr Lys Gln Cys Ala 85 90
95Trp Asn Ser Leu Asn Tyr Gly Ile Asp Leu Gly Lys Ile Ala Asp Cys
100 105 110Thr Phe Thr Lys Leu
Arg Ser Glu Ser Ala Leu Arg Val Leu Phe Thr 115
120 125Gly Ser Leu Arg Leu Lys Cys Lys Glu Ala Cys Cys
Gln Arg Trp Tyr 130 135 140Phe Thr Phe
Asp Gly Ala Glu Cys Thr Gly Pro Leu Pro Val Glu Ser145
150 155 160Ile Ile Tyr Leu Asn Gln Gly
Ser Pro Glu Leu Asn Ser Thr Ile Asn 165
170 175Ile His Arg Thr Ser Ser Val Glu Gly Leu Cys Glu
Gly Ile Lys Ala 180 185 190Gly
Leu Val Asp Val Ala Leu Trp Val Gly Thr Cys Ala Asp Tyr Pro 195
200 205Arg Gly Asp Ala Ser Thr Gly Trp Asn
Ser Val Ser Arg Ile Ile Ile 210 215
220Glu Glu Leu Pro Lys2256231PRTDanio rerio 6Met Gly Thr Lys Leu Thr Gln
Leu Leu Ile Cys Phe Trp Ile Ser Leu1 5 10
15Pro Phe Cys Val Thr Gln Lys Ala Lys Glu Arg Ile Pro
Arg Gln Arg 20 25 30Asp Ala
Glu Phe Thr Asp Lys Tyr Gln Ala Cys Val Gln Gly Val Pro 35
40 45Gly Val Gln Gly Arg Asp Gly Asn Pro Gly
Ile Asn Gly Ile Pro Gly 50 55 60Thr
Pro Gly Ile Pro Gly Arg Asp Gly Leu Lys Gly Glu Lys Gly Glu65
70 75 80Cys Val Ser Glu Arg Phe
Glu Glu Pro Trp Lys Pro Asn Phe Lys Gln 85
90 95Cys Ala Trp Asn Ser Leu Asn Tyr Gly Ile Asp Leu
Gly Lys Ile Ala 100 105 110Glu
Cys Thr Phe Thr Lys Gln Arg Ser Asp Ser Ala Leu Arg Val Leu 115
120 125Phe Ser Gly Ser Leu Arg Leu Lys Cys
Lys Thr Ala Cys Cys Gln Arg 130 135
140Trp Tyr Phe Thr Phe Asn Gly Ala Glu Cys Thr Gly Pro Leu Pro Ile145
150 155 160Glu Ser Ile Val
Tyr Leu Asp Gln Gly Ser Pro Glu Leu Asn Ser Thr 165
170 175Ile Asn Ile His Arg Thr Ser Thr Val Glu
Gly Leu Cys Glu Gly Ile 180 185
190His Ala Gly Leu Val Asp Val Gly Ile Trp Val Gly Thr Cys Ala Asp
195 200 205Tyr Pro Arg Gly Asp Ala Ser
Thr Gly Trp Asn Ser Val Ser Arg Val 210 215
220Ile Ile Glu Glu Leu Pro Lys225 2307201PRTGallus
gallus 7Arg Pro Arg Glu Val Leu Glu Ala Tyr Asn Gly Val Cys Leu Gln Gly1
5 10 15Pro Ser Gly Val
Pro Gly Arg Asp Gly Asn Pro Gly Thr Asn Gly Ile 20
25 30Pro Gly Thr Pro Gly Ile Pro Gly Arg Asp Gly
Pro Lys Gly Glu Lys 35 40 45Gly
Glu Cys Leu Arg Glu Ser Ile Glu Glu Ser Trp Thr Pro Asn Phe 50
55 60Lys Gln Cys Ser Trp Ser Ala Leu Asn Tyr
Gly Ile Asp Leu Gly Lys65 70 75
80Ile Ala Glu Cys Thr Phe Thr Lys Met Arg Ser Asn Ser Ala Leu
Arg 85 90 95Val Leu Phe
Ser Gly Ser Leu Arg Leu Lys Cys Arg Ser Ala Cys Cys 100
105 110Gln Arg Trp Tyr Phe Thr Phe Asn Gly Ala
Glu Cys Ala Gly Pro Leu 115 120
125Pro Ile Glu Ala Ile Ile Tyr Leu Asp Gln Gly Ser Pro Glu Leu Asn 130
135 140Ser Thr Ile Asn Ile His Arg Thr
Ser Ser Val Glu Gly Leu Cys Glu145 150
155 160Gly Ile Asn Ala Gly Leu Val Asp Ile Ala Ile Trp
Val Gly Thr Cys 165 170
175Ser Asp Tyr Pro Arg Gly Asp Ala Ser Thr Gly Trp Asn Ser Val Ser
180 185 190Arg Ile Ile Ile Glu Glu
Leu Pro Lys 195 20086PRTArtificial
SequenceDescription of Artificial Sequence Synthetic peptide 8Thr
Ile Ser Gly Ser Asp1 5911PRTArtificial SequenceDescription
of Artificial Sequence Synthetic peptide 9Gly Arg Ile Ser Pro Tyr
Gly Gly Asn Thr Asn1 5
101013PRTArtificial SequenceDescription of Artificial Sequence Synthetic
peptide 10Cys Ala Arg Val Gly Gly Leu Lys Leu Leu Phe Asp Tyr1
5 10116PRTArtificial SequenceDescription of
Artificial Sequence Synthetic peptide 11Thr Ile Thr Asn Ser Asp1
51211PRTArtificial SequenceDescription of Artificial Sequence
Synthetic peptide 12Ala Thr Ile Tyr Pro Tyr Gly Gly Tyr Thr Tyr1
5 101314PRTArtificial SequenceDescription of
Artificial Sequence Synthetic peptide 13Cys Ala Arg Gly Gly Gly Met
Asp Gly Tyr Val Met Asp Tyr1 5
10146PRTArtificial SequenceDescription of Artificial Sequence Synthetic
peptide 14Thr Ile Asn Asn Tyr Asp1 51511PRTArtificial
SequenceDescription of Artificial Sequence Synthetic peptide 15Gly
Tyr Ile Ser Pro Pro Ser Gly Ala Thr Tyr1 5
101614PRTArtificial SequenceDescription of Artificial Sequence Synthetic
peptide 16Cys Ala Arg Met Val Gly Met Arg Arg Gly Val Met Asp Tyr1
5 10176PRTArtificial SequenceDescription of
Artificial Sequence Synthetic peptide 17Thr Ile Ser Gly Ser Trp1
51811PRTArtificial SequenceDescription of Artificial Sequence
Synthetic peptide 18Ala Trp Ile Ala Pro Tyr Ser Gly Ala Thr Asp1
5 101913PRTArtificial SequenceDescription of
Artificial Sequence Synthetic peptide 19Cys Ala Arg Glu Gly Gly Leu
Tyr Trp Val Phe Asp Tyr1 5
10206PRTArtificial SequenceDescription of Artificial Sequence Synthetic
peptide 20Thr Ile Ser Asn Tyr Gly1 52111PRTArtificial
SequenceDescription of Artificial Sequence Synthetic peptide 21Gly
Arg Ile Ser Pro Ser Asn Gly Ser Thr Tyr1 5
102210PRTArtificial SequenceDescription of Artificial Sequence Synthetic
peptide 22Cys Ala Lys Cys Ser Val Arg Phe Ala Tyr1 5
102330DNAArtificial SequenceDescription of Artificial
Sequence Synthetic oligonucleotide 23catccagtag aagcatctcc
ttttgggtaa 302419DNAArtificial
SequenceDescription of Artificial Sequence Synthetic oligonucleotide
24gggttggcac ttgttcaga
192524DNAArtificial SequenceDescription of Artificial Sequence Synthetic
oligonucleotide 25caataatgat gcgagaaact gaat
2426266DNAArtificial SequenceDescription of Artificial
Sequence Synthetic polynucleotide 26gggagccatg cgaccccagg gccccgccgc
ctccccgcag cggctccgcg gcctcctgct 60gctcctgctg ctgcagctgc ccgcgccgtc
gagcgcctct gagatcccca aggggaagca 120aaaggcgcag ctccggcaga gggaggtggt
ggacctgtat aatggaatgt gcttacaagg 180gccagcagga gtgcctggtc gagacgggag
ccctggggcc aatgttattc cgggtacacc 240tgggatccca ggtcgggatg gattca
2662744DNAArtificial
SequenceDescription of Artificial Sequence Synthetic oligonucleotide
27gatcgcggcc gcacaccacc atcaccatca ccatcactaa gtga
442844DNAArtificial SequenceDescription of Artificial Sequence Synthetic
oligonucleotide 28ggcctcactt agtgatggtg atggtgatgg tggtgtgcgg ccgc
442941DNAArtificial SequenceDescription of Artificial
Sequence Synthetic oligonucleotide 29ctgcgctagc accatgatgg
gtactaaact gactcaactt t 413035DNAArtificial
SequenceDescription of Artificial Sequence Synthetic oligonucleotide
30ggaagcggcc gcttttggaa gctcttcaat gatca
353155DNAArtificial SequenceDescription of Artificial Sequence Synthetic
oligonucleotide 31gaagctagca ccatgcaccc ccaaggccgc gcggcccccc
cgcagctgct gctcg 553238DNAArtificial SequenceDescription of
Artificial Sequence Synthetic oligonucleotide 32gaagcggccg
cttatttcgg tagttcttca atgatgat
383337DNAArtificial SequenceDescription of Artificial Sequence Synthetic
oligonucleotide 33ccagctagca ccatgcgacc ccagggcccc gccgcct
373438DNAArtificial SequenceDescription of Artificial
Sequence Synthetic oligonucleotide 34caagcggccg cttattttgg
tagttcttca ataatgat 383535DNAArtificial
SequenceDescription of Artificial Sequence Synthetic oligonucleotide
35caagcggccg cttttggtag ttcttcaata atgat
353643DNAArtificial SequenceDescription of Artificial Sequence Synthetic
oligonucleotide 36tcaaagcttt ccctcagcat tccattatac aggtccacca cct
433742DNAArtificial SequenceDescription of Artificial
Sequence Synthetic oligonucleotide 37ctcaaagctt tccctcagat
acaggtccac cacctccctc tg 423832DNAArtificial
SequenceDescription of Artificial Sequence Synthetic oligonucleotide
38ggggagctca gaggcgctcg acggcgcggg ca
323930DNAArtificial SequenceDescription of Artificial Sequence Synthetic
oligonucleotide 39acaggtcgac cacctccctc tgccggagct
304034DNAArtificial SequenceDescription of Artificial
Sequence Synthetic oligonucleotide 40gaagagctca gggaaagctt
tgaggagtcc tgga 344145DNAArtificial
SequenceDescription of Artificial Sequence Synthetic oligonucleotide
41gtggtcgacc tgtataatgc aatgtgctta caagggccag cagga
454236DNAArtificial SequenceDescription of Artificial Sequence Synthetic
oligonucleotide 42aattgtcgac gccatttcag ggcttccttg gtccaa
364330DNAArtificial SequenceDescription of Artificial
Sequence Synthetic oligonucleotide 43tggcgtcgac aattaatatt
catcgcactt 304451DNAArtificial
SequenceDescription of Artificial Sequence Synthetic oligonucleotide
44gtggtcgacc tgtataatgg aatggcctta caagggccag caggagtgcc t
514543DNAArtificial SequenceDescription of Artificial Sequence Synthetic
oligonucleotide 45ctcaaagctt tccctcagag cttccccctt ttctcctttg aat
434628DNAArtificial SequenceDescription of Artificial
Sequence Synthetic oligonucleotide 46ccatgaggcc tgcttgtagt tgggtgtc
284729DNAArtificial
SequenceDescription of Artificial Sequence Synthetic oligonucleotide
47agcaggcctc atggagttca ttgaattat
294828DNAArtificial SequenceDescription of Artificial Sequence Synthetic
oligonucleotide 48taaacgttgc ctccgcaatt ttcccaag
284927DNAArtificial SequenceDescription of Artificial
Sequence Synthetic oligonucleotide 49ggcaacgttt acaaagatgc gttcaaa
275034DNAArtificial
SequenceDescription of Artificial Sequence Synthetic oligonucleotide
50gcatgcattt ctggctttta gccgaagtga gcca
345126DNAArtificial SequenceDescription of Artificial Sequence Synthetic
oligonucleotide 51ctggcatgct gcatttctgc atttta
265232DNAArtificial SequenceDescription of Artificial
Sequence Synthetic oligonucleotide 52gcagcatgcc agcgttggta
tttcacattc aa 325330DNAArtificial
SequenceDescription of Artificial Sequence Synthetic oligonucleotide
53aatgcatgcg ctcagcgttg gtatttcaca
305430DNAArtificial SequenceDescription of Artificial Sequence Synthetic
oligonucleotide 54cctgaggcct cagctccatt gaatgtgaaa
305529DNAArtificial SequenceDescription of Artificial
Sequence Synthetic oligonucleotide 55ctgaggcctc aggacctctt cccattgaa
295650DNAArtificial
SequenceDescription of Artificial Sequence Synthetic oligonucleotide
56tccggcgcca attccttcag caagtccttc cacagaagaa gtgcgatgaa
505731DNAArtificial SequenceDescription of Artificial Sequence Synthetic
oligonucleotide 57attggcgccg gattagtgga tgttgctatc t
315830DNAArtificial SequenceDescription of Artificial
Sequence Synthetic oligonucleotide 58tgaagcggta ccaacccaga
tagcaacatc 305932DNAArtificial
SequenceDescription of Artificial Sequence Synthetic oligonucleotide
59ggcggtaccg cttcagatta cccaaaagga ga
326042DNAArtificial SequenceDescription of Artificial Sequence Synthetic
oligonucleotide 60gtggtcgacc tgtatgctgg aatgtgctta caagggccag ca
426147DNAArtificial SequenceDescription of Artificial
Sequence Synthetic oligonucleotide 61gtggtcgacc tgtataatgg
agcgtgctta caagggccag caggagt 476230DNAArtificial
SequenceDescription of Artificial Sequence Synthetic oligonucleotide
62tcggccggcc actcctgctg gcccttgtaa
306346DNAArtificial SequenceDescription of Artificial Sequence Synthetic
oligonucleotide 63gtggccggcc gagacgggag cgctggggcc aatggcattc cgggta
466427DNAArtificial SequenceDescription of Artificial
Sequence Synthetic oligonucleotide 64tgtgccggca atgccattgg ccccagg
276550DNAArtificial
SequenceDescription of Artificial Sequence Synthetic oligonucleotide
65attgccggca cagctgggat cgcaggtcgg gatggattca aaggagaaaa
506630DNAArtificial SequenceDescription of Artificial Sequence Synthetic
oligonucleotide 66ttcgccggcg aatccatccc gacctgggat
306739DNAArtificial SequenceDescription of Artificial
Sequence Synthetic oligonucleotide 67ttcgccggcg aagcggggga
atgtctgagg gaaagcttt 396832DNAArtificial
SequenceDescription of Artificial Sequence Synthetic oligonucleotide
68gaagcggccg ctttcggtag ttcttcaatg at
326935DNAArtificial SequenceDescription of Artificial Sequence Synthetic
oligonucleotide 69gaagcggccg caagacttgg gtaaaaatcg tattt
357041DNAArtificial SequenceDescription of Artificial
Sequence Synthetic oligonucleotide 70gaaagatctc tagtctgaca
aagagaaaca ctgctttagg a 417125DNAArtificial
SequenceDescription of Artificial Sequence Synthetic oligonucleotide
71ggatcgtcgg ttttgtacaa tatgt
257230DNAArtificial SequenceDescription of Artificial Sequence Synthetic
oligonucleotide 72ggggatctca gacgcaaagg cagaatgcgc
307330DNAArtificial SequenceDescription of Artificial
Sequence Synthetic oligonucleotide 73tctgcctttg cgtctgagat
ccccaagggg 307439DNAArtificial
SequenceDescription of Artificial Sequence Synthetic oligonucleotide
74ccgttctgca gttaatgatg atgatgatga tgatgatgg
397530DNAArtificial SequenceDescription of Artificial Sequence Synthetic
oligonucleotide 75gctttccctc agcgcaaagg cagaatgcgc
307631DNAArtificial SequenceDescription of Artificial
Sequence Synthetic oligonucleotide 76tctgcctttg cgctgaggga
aagctttgag g 317732DNAArtificial
SequenceDescription of Artificial Sequence Synthetic oligonucleotide
77ccgggatcct taatgatgat gatgatgatg at
327831DNAArtificial SequenceDescription of Artificial Sequence Synthetic
oligonucleotide 78attccccctt ttccgcaaag gcagaatgcg c
317932DNAArtificial SequenceDescription of Artificial
Sequence Synthetic oligonucleotide 79tctgcctttg cggaaaaggg
ggaatgtctg ag 328023DNAArtificial
SequenceDescription of Artificial Sequence Synthetic oligonucleotide
80atcgccaatg ccaactcccg tca
238123DNAArtificial SequenceDescription of Artificial Sequence Synthetic
oligonucleotide 81gcttgcgtgc ttccttggtc tta
238228DNAArtificial SequenceDescription of Artificial
Sequence Synthetic oligonucleotide 82tgctgctgca gctgcccgcg ccgtcgag
288328DNAArtificial
SequenceDescription of Artificial Sequence Synthetic oligonucleotide
83tccagtagaa gcatctcctt ttgggtaa
288435PRTSaccharomyces cerevisiae 84Gly Arg Met Lys Gln Leu Glu Asp Lys
Val Glu Glu Leu Leu Ser Lys1 5 10
15Asn Tyr His Leu Glu Asn Glu Val Ala Arg Leu Lys Lys Leu Val
Gly 20 25 30Glu Arg Gly
358528DNAArtificial SequenceDescription of Artificial Sequence
Synthetic oligonucleotide 85gctcggttgc cgccgggcgt tttttatg
2886255PRTArtificial SequenceDescription of
Artificial Sequence Synthetic polypeptide 86Met Lys Lys Asn Ile Ala
Phe Leu Leu Ala Ser Met Phe Val Phe Ser1 5
10 15Ile Ala Thr Asn Ala Tyr Ala Asp Ile Gln Met Thr
Gln Ser Pro Ser 20 25 30Ser
Leu Ser Ala Ser Val Gly Asp Arg Val Thr Ile Thr Cys Arg Ala 35
40 45Ser Gln Asp Val Ser Thr Ala Val Ala
Trp Tyr Gln Gln Lys Pro Gly 50 55
60Lys Ala Pro Lys Leu Leu Ile Tyr Ser Ala Ser Phe Leu Tyr Ser Gly65
70 75 80Val Pro Ser Arg Phe
Ser Gly Ser Gly Ser Gly Thr Asp Phe Thr Leu 85
90 95Thr Ile Ser Ser Leu Gln Pro Glu Asp Phe Ala
Thr Tyr Tyr Cys Gln 100 105
110Gln Ser Tyr Thr Thr Pro Pro Thr Phe Gly Gln Gly Thr Lys Val Glu
115 120 125Ile Lys Arg Thr Val Ala Ala
Pro Ser Val Phe Ile Phe Pro Pro Ser 130 135
140Asp Glu Gln Leu Lys Ser Gly Thr Ala Ser Val Val Cys Leu Leu
Asn145 150 155 160Asn Phe
Tyr Pro Arg Glu Ala Lys Val Gln Trp Lys Val Asp Asn Ala
165 170 175Leu Gln Ser Gly Asn Ser Gln
Glu Ser Val Thr Glu Gln Asp Ser Lys 180 185
190Asp Ser Thr Tyr Ser Leu Ser Ser Thr Leu Thr Leu Ser Lys
Ala Asp 195 200 205Tyr Glu Lys His
Lys Val Tyr Ala Cys Glu Val Thr His Gln Gly Leu 210
215 220Ser Ser Pro Val Thr Lys Ser Phe Asn Arg Gly Glu
Cys Gly Ala Ser225 230 235
240Ser Gly Met Ala Asp Pro Asn Arg Phe Arg Gly Lys Asp Leu Ala
245 250 25587409PRTArtificial
SequenceDescription of Artificial Sequence Synthetic polypeptide
87Met Lys Lys Asn Ile Ala Phe Leu Leu Ala Ser Met Phe Val Phe Ser1
5 10 15Ile Ala Thr Asn Ala Tyr
Ala Glu Val Gln Leu Val Glu Ser Gly Gly 20 25
30Gly Leu Val Gln Pro Gly Gly Ser Leu Arg Leu Ser Cys
Ala Ala Ser 35 40 45Gly Phe Asn
Ile Lys Asp Thr Tyr Ile His Trp Val Arg Gln Ala Pro 50
55 60Gly Lys Gly Leu Glu Trp Val Ala Arg Ile Tyr Pro
Thr Asn Gly Tyr65 70 75
80Thr Arg Tyr Ala Asp Ser Val Lys Gly Arg Phe Thr Ile Ser Ala Asp
85 90 95Thr Ser Lys Asn Thr Ala
Tyr Leu Gln Met Asn Ser Leu Arg Ala Glu 100
105 110Asp Thr Ala Val Tyr Tyr Cys Ser Arg Trp Gly Gly
Asp Gly Phe Tyr 115 120 125Ala Met
Asp Tyr Trp Gly Gln Gly Thr Leu Val Thr Val Ser Ser Ala 130
135 140Ser Thr Lys Gly Pro Ser Val Phe Pro Leu Ala
Pro Ser Ser Lys Ser145 150 155
160Thr Ser Gly Gly Thr Ala Ala Leu Gly Cys Leu Val Lys Asp Tyr Phe
165 170 175Pro Glu Pro Val
Thr Val Ser Trp Asn Ser Gly Ala Leu Thr Ser Gly 180
185 190Val His Thr Phe Pro Ala Val Leu Gln Ser Ser
Gly Leu Tyr Ser Leu 195 200 205Ser
Ser Val Val Thr Val Pro Ser Ser Ser Leu Gly Thr Gln Thr Tyr 210
215 220Ile Cys Asn Val Asn His Lys Pro Ser Asn
Thr Lys Val Asp Lys Lys225 230 235
240Val Glu Pro Lys Ser Cys Asp Lys Thr His Leu Ser Gly Gly Gly
Ser 245 250 255Gly Ser Gly
Asp Phe Asp Tyr Glu Lys Met Ala Asn Ala Asn Lys Gly 260
265 270Ala Met Thr Glu Asn Ala Asp Glu Asn Ala
Leu Gln Ser Asp Ala Lys 275 280
285Gly Lys Leu Asp Ser Val Ala Thr Asp Tyr Gly Ala Ala Ile Asp Gly 290
295 300Phe Ile Gly Asp Val Ser Gly Leu
Ala Asn Gly Asn Gly Ala Thr Gly305 310
315 320Asp Phe Ala Gly Ser Asn Ser Gln Met Ala Gln Val
Gly Asp Gly Asp 325 330
335Asn Ser Pro Leu Met Asn Asn Phe Arg Gln Tyr Leu Pro Ser Leu Pro
340 345 350Gln Ser Val Glu Cys Arg
Pro Phe Val Phe Ser Ala Gly Lys Pro Tyr 355 360
365Glu Phe Ser Ile Asp Cys Asp Lys Ile Asn Leu Phe Arg Gly
Val Phe 370 375 380Ala Phe Leu Leu Tyr
Val Ala Thr Phe Met Tyr Val Phe Ser Thr Phe385 390
395 400Ala Asn Ile Leu Arg Asn Lys Glu Ser
405887060DNAArtificial SequenceDescription of Artificial
Sequence Synthetic polynucleotide 88gaattcaact tctccatact ttggataagg
aaatacagac atgaaaaatc tcattgctga 60gttgttattt aagcttgccc aaaaagaaga
agagtcgaat gaactgtgtg cgcaggtaga 120agctttggag attatcgtca ctgcaatgct
tcgcaatatg gcgcaaaatg accaacagcg 180gttgattgat caggtagagg gggcgctgta
cgaggtaaag cccgatgcca gcattcctga 240cgacgatacg gagctgctgc gcgattacgt
aaagaagtta ttgaagcatc ctcgtcagta 300aaaagttaat cttttcaaca gctgtcataa
agttgtcacg gccgagactt atagtcgctt 360tgtttttatt ttttaatgta tttgtaacta
gtacgcaagt tcacgtaaaa agggtatgta 420gaggttgagg tgattttatg aaaaagaata
tcgcatttct tcttgcatct atgttcgttt 480tttctattgc tacaaatgcc tatgcagata
tccagatgac ccagtccccg agctccctgt 540ccgcctctgt gggcgatagg gtcaccatca
cctgccgtgc cagtcaggat gtgtccactg 600ctgtagcctg gtatcaacag aaaccaggaa
aagctccgaa gcttctgatt tactcggcat 660ccttcctcta ctctggagtc ccttctcgct
tctctggtag cggttccggg acggatttca 720ctctgaccat cagcagtctg cagccggaag
acttcgcaac ttattactgt cagcaatctt 780atactactcc tcccacgttc ggacagggta
ccaaggtgga gatcaaacga actgtggctg 840caccatctgt cttcatcttc ccgccatctg
atgagcagtt gaaatctgga actgcctctg 900ttgtgtgcct gctgaataac ttctatccca
gagaggccaa agtacagtgg aaggtggata 960acgccctcca atcgggtaac tcccaggaga
gtgtcacaga gcaggacagc aaggacagca 1020cctacagcct cagcagcacc ctgacgctga
gcaaagcaga ctacgagaaa cacaaagtct 1080acgcctgcga agtcacccat cagggcctga
gctcgcccgt cacaaagagc ttcaacaggg 1140gagagtgtgg tgccagctcc ggtatggctg
atccgaaccg tttccgcggt aaggacctgg 1200cataactcga ggctgatcct ctacgccgga
cgcatcgtgg ccctagtacg caagttcacg 1260taaaaagggt aactagaggt tgaggtgatt
ttatgaaaaa gaatatcgca tttcttcttg 1320catctatgtt cgttttttct attgctacaa
acgcgtacgc tgaggttcag ctggtggagt 1380ctggcggtgg cctggtgcag ccagggggct
cactccgttt gtcctgtgca gcttctggct 1440tcaacattaa agacacctat atacactggg
tgcgtcaggc cccgggtaag ggcctggaat 1500gggttgcaag gatttatcct acgaatggtt
atactagata tgccgatagc gtcaagggcc 1560gtttcactat aagcgcagac acatccaaaa
acacagccta cctacaaatg aacagcttaa 1620gagctgagga cactgccgtc tattattgta
gccgctgggg aggggacggc ttctatgcta 1680tggactactg gggtcaagga acactagtca
ccgtctcctc ggcctccacc aagggcccat 1740cggtcttccc cctggcaccc tcctccaaga
gcacctctgg gggcacagcg gccctgggct 1800gcctggtcaa ggactacttc cccgaaccgg
tgacggtgtc gtggaactca ggcgccctga 1860ccagcggcgt gcacaccttc ccggctgtcc
tacagtcctc aggactctac tccctcagca 1920gcgtggtgac cgtgccctcc agcagcttgg
gcacccagac ctacatctgc aacgtgaatc 1980acaagcccag caacaccaag gtcgacaaga
aagttgagcc caaatcttgt gacaaaactc 2040acctcagtgg cggtggctct ggttccggtg
attttgatta tgaaaagatg gcaaacgcta 2100ataagggggc tatgaccgaa aatgccgatg
aaaacgcgct acagtctgac gctaaaggca 2160aacttgattc tgtcgctact gattacggtg
ctgctatcga tggtttcatt ggtgacgttt 2220ccggccttgc taatggtaat ggtgctactg
gtgattttgc tggctctaat tcccaaatgg 2280ctcaagtcgg tgacggtgat aattcacctt
taatgaataa tttccgtcaa tatttacctt 2340ccctccctca atcggttgaa tgtcgccctt
ttgtctttag cgctggtaaa ccatatgaat 2400tttctattga ttgtgacaaa ataaacttat
tccgtggtgt ctttgcgttt cttttatatg 2460ttgccacctt tatgtatgta ttttctacgt
ttgctaacat actgcgtaat aaggagtctt 2520aatcatgcca gttcttttgg ctagcgccgc
cctatacctt gtctgcctcc ccgcgttgcg 2580tcgcggtgca tggagccggg ccacctcgac
ctgaatggaa gccggcggca cctcgctaac 2640ggattcacca ctccaagaat tggagccaat
caattcttgc ggagaactgt gaatgcgcaa 2700accaaccctt ggcagaacat atccatcgcg
tccgccatct ccagcagccg cacgcggcgc 2760atctcgggca gcgttgggtc ctggccacgg
gtgcgcatga tcgtgctcct gtcgttgagg 2820acccggctag gctggcgggg ttgccttact
ggttagcaga atgaatcacc gatacgcgag 2880cgaacgtgaa gcgactgctg ctgcaaaacg
tctgcgacct gagcaacaac atgaatggtc 2940ttcggtttcc gtgtttcgta aagtctggaa
acgcggaagt cagcgccctg caccattatg 3000ttccggatct gcatcgcagg atgctgctgg
ctaccctgtg gaacacctac atctgtatta 3060acgaagcgct ggcattgacc ctgagtgatt
tttctctggt cccgccgcat ccataccgcc 3120agttgtttac cctcacaacg ttccagtaac
cgggcatgtt catcatcagt aacccgtatc 3180gtgagcatcc tctctcgttt catcggtatc
attaccccca tgaacagaaa ttccccctta 3240cacggaggca tcaagtgacc aaacaggaaa
aaaccgccct taacatggcc cgctttatca 3300gaagccagac attaacgctt ctggagaaac
tcaacgagct ggacgcggat gaacaggcag 3360acatctgtga atcgcttcac gaccacgctg
atgagcttta ccgcaggatc cggaaattgt 3420aaacgttaat attttgttaa aattcgcgtt
aaatttttgt taaatcagct cattttttaa 3480ccaataggcc gaaatcggca aaatccctta
taaatcaaaa gaatagaccg agatagggtt 3540gagtgttgtt ccagtttgga acaagagtcc
actattaaag aacgtggact ccaacgtcaa 3600agggcgaaaa accgtctatc agggctatgg
cccactacgt gaaccatcac cctaatcaag 3660ttttttgggg tcgaggtgcc gtaaagcact
aaatcggaac cctaaaggga gcccccgatt 3720tagagcttga cggggaaagc cggcgaacgt
ggcgagaaag gaagggaaga aagcgaaagg 3780agcgggcgct agggcgctgg caagtgtagc
ggtcacgctg cgcgtaacca ccacacccgc 3840cgcgcttaat gcgccgctac agggcgcgtc
cggatcctgc ctcgcgcgtt tcggtgatga 3900cggtgaaaac ctctgacaca tgcagctccc
ggagacggtc acagcttgtc tgtaagcgga 3960tgccgggagc agacaagccc gtcagggcgc
gtcagcgggt gttggcgggt gtcggggcgc 4020agccatgacc cagtcacgta gcgatagcgg
agtgtatact ggcttaacta tgcggcatca 4080gagcagattg tactgagagt gcaccatatg
cggtgtgaaa taccgcacag atgcgtaagg 4140agaaaatacc gcatcaggcg ctcttccgct
tcctcgctca ctgactcgct gcgctcggtc 4200gttcggctgc ggcgagcggt atcagctcac
tcaaaggcgg taatacggtt atccacagaa 4260tcaggggata acgcaggaaa gaacatgtga
gcaaaaggcc agcaaaaggc caggaaccgt 4320aaaaaggccg cgttgctggc gtttttccat
aggctccgcc cccctgacga gcatcacaaa 4380aatcgacgct caagtcagag gtggcgaaac
ccgacaggac tataaagata ccaggcgttt 4440ccccctggaa gctccctcgt gcgctctcct
gttccgaccc tgccgcttac cggatacctg 4500tccgcctttc tcccttcggg aagcgtggcg
ctttctcata gctcacgctg taggtatctc 4560agttcggtgt aggtcgttcg ctccaagctg
ggctgtgtgc acgaaccccc cgttcagccc 4620gaccgctgcg ccttatccgg taactatcgt
cttgagtcca acccggtaag acacgactta 4680tcgccactgg cagcagccac tggtaacagg
attagcagag cgaggtatgt aggcggtgct 4740acagagttct tgaagtggtg gcctaactac
ggctacacta gaaggacagt atttggtatc 4800tgcgctctgc tgaagccagt taccttcgga
aaaagagttg gtagctcttg atccggcaaa 4860caaaccaccg ctggtagcgg tggttttttt
gtttgcaagc agcagattac gcgcagaaaa 4920aaaggatctc aagaagatcc tttgatcttt
tctacggggt ctgacgctca gtggaacgaa 4980aactcacgtt aagggatttt ggtcatgaga
ttatcaaaaa ggatcttcac ctagatcctt 5040ttaaattaaa aatgaagttt taaatcaatc
taaagtatat atgagtaaac ttggtctgac 5100agttaccaat gcttaatcag tgaggcacct
atctcagcga tctgtctatt tcgttcatcc 5160atagttgcct gactccccgt cgtgtagata
actacgatac gggagggctt accatctggc 5220cccagtgctg caatgatacc gcgagaccca
cgctcaccgg ctccagattt atcagcaata 5280aaccagccag ccggaagggc cgagcgcaga
agtggtcctg caactttatc cgcctccatc 5340cagtctatta attgttgccg ggaagctaga
gtaagtagtt cgccagttaa tagtttgcgc 5400aacgttgttg ccattgctgc aggcatcgtg
gtgtcacgct cgtcgtttgg tatggcttca 5460ttcagctccg gttcccaacg atcaaggcga
gttacatgat cccccatgtt gtgcaaaaaa 5520gcggttagct ccttcggtcc tccgatcgtt
gtcagaagta agttggccgc agtgttatca 5580ctcatggtta tggcagcact gcataattct
cttactgtca tgccatccgt aagatgcttt 5640tctgtgactg gtgagtactc aaccaagtca
ttctgagaat agtgtatgcg gcgaccgagt 5700tgctcttgcc cggcgtcaac acgggataat
accgcgccac atagcagaac tttaaaagtg 5760ctcatcattg gaaaacgttc ttcggggcga
aaactctcaa ggatcttacc gctgttgaga 5820tccagttcga tgtaacccac tcgtgcaccc
aactgatctt cagcatcttt tactttcacc 5880agcgtttctg ggtgagcaaa aacaggaagg
caaaatgccg caaaaaaggg aataagggcg 5940acacggaaat gttgaatact catactcttc
ctttttcaat attattgaag catttatcag 6000ggttattgtc tcatgagcgg atacatattt
gaatgtattt agaaaaataa acaaataggg 6060gttccgcgca catttccccg aaaagtgcca
cctgacgtct aagaaaccat tattatcatg 6120acattaacct ataaaaatag gcgtatcacg
aggccctttc gtcttcaata caggtagacc 6180tttcgtagag atgtacagtg aaatccccga
aattatacac atgactgaag gaagggagct 6240cgtcattccc tgccgggtta cgtcacctaa
catcactgtt actttaaaaa agtttccact 6300tgacactttg atccctgatg gaaaacgcat
aatctgggac agtagaaagg gcttcatcat 6360atcaaatgca acgtacaaag aaatagggct
tctgacctgt gaagcaacag tcaatgggca 6420tttgtataag acaaactatc tcacacatcg
acaaaccaat acaatacagg tagacctttc 6480gtagagatgt acagtgaaat ccccgaaatt
atacacatga ctgaaggaag ggagctcgtc 6540attccctgcc gggttacgtc acctaacatc
actgttactt taaaaaagtt tccacttgac 6600actttgatcc ctgatggaaa acgcataatc
tgggacagta gaaagggctt catcatatca 6660aatgcaacgt acaaagaaat agggcttctg
acctgtgaag caacagtcaa tgggcatttg 6720tataagacaa actatctcac acatcgacaa
accaatacaa tctacaggta gacctttcgt 6780agagatgtac agtgaaatcc ccgaaattat
acacatgact gaaggaaggg agctcgtcat 6840tccctgccgg gttacgtcac ctaacatcac
tgttacttta aaaaagtttc cacttgacac 6900tttgatccct gatggaaaac gcataatctg
ggacagtaga aagggcttca tcatatcaaa 6960tgcaacgtac aaagaaatag ggcttctgac
ctgtgaagca acagtcaatg ggcatttgta 7020taagacaaac tatctcacac atcgacaaac
caatacaatc 706089256PRTArtificial
SequenceDescription of Artificial Sequence Synthetic polypeptide
89Met Lys Lys Asn Ile Ala Phe Leu Leu Ala Ser Met Phe Val Phe Ser1
5 10 15Ile Ala Thr Asn Ala Tyr
Ala Ser Asp Ile Gln Met Thr Gln Ser Pro 20 25
30Ser Ser Leu Ser Ala Ser Val Gly Asp Arg Val Thr Ile
Thr Cys Arg 35 40 45Ala Ser Gln
Asp Val Ser Thr Ala Val Ala Trp Tyr Gln Gln Lys Pro 50
55 60Gly Lys Ala Pro Lys Leu Leu Ile Tyr Ser Ala Ser
Phe Leu Tyr Ser65 70 75
80Gly Val Pro Ser Arg Phe Ser Gly Ser Gly Ser Gly Thr Asp Phe Thr
85 90 95Leu Thr Ile Ser Ser Leu
Gln Pro Glu Asp Phe Ala Thr Tyr Tyr Cys 100
105 110Gln Gln Ser Tyr Thr Thr Pro Pro Thr Phe Gly Gln
Gly Thr Lys Val 115 120 125Glu Ile
Lys Arg Thr Val Ala Ala Pro Ser Val Phe Ile Phe Pro Pro 130
135 140Ser Asp Glu Gln Leu Lys Ser Gly Thr Ala Ser
Val Val Cys Leu Leu145 150 155
160Asn Asn Phe Tyr Pro Arg Glu Ala Lys Val Gln Trp Lys Val Asp Asn
165 170 175Ala Leu Gln Ser
Gly Asn Ser Gln Glu Ser Val Thr Glu Gln Asp Ser 180
185 190Lys Asp Ser Thr Tyr Ser Leu Ser Ser Thr Leu
Thr Leu Ser Lys Ala 195 200 205Asp
Tyr Glu Lys His Lys Val Tyr Ala Cys Glu Val Thr His Gln Gly 210
215 220Leu Ser Ser Pro Val Thr Lys Ser Phe Asn
Arg Gly Glu Cys Gly Ala225 230 235
240Ser Ser Gly Met Ala Asp Pro Asn Arg Phe Arg Gly Lys Asp Leu
Ala 245 250
25590445PRTArtificial SequenceDescription of Artificial Sequence
Synthetic polypeptide 90Met Lys Lys Asn Ile Ala Phe Leu Leu Ala Ser
Met Phe Val Phe Ser1 5 10
15Ile Ala Thr Asn Ala Tyr Ala Glu Val Gln Leu Val Glu Ser Gly Gly
20 25 30Gly Leu Val Gln Pro Gly Gly
Ser Leu Arg Leu Ser Cys Ala Ala Ser 35 40
45Gly Phe Asn Ile Lys Asp Thr Tyr Ile His Trp Val Arg Gln Ala
Pro 50 55 60Gly Lys Gly Leu Glu Trp
Val Ala Arg Ile Tyr Pro Thr Asn Gly Tyr65 70
75 80Thr Arg Tyr Ala Asp Ser Val Lys Gly Arg Phe
Thr Ile Ser Ala Asp 85 90
95Thr Ser Lys Asn Thr Ala Tyr Leu Gln Met Asn Ser Leu Arg Ala Glu
100 105 110Asp Thr Ala Val Tyr Tyr
Cys Ser Arg Trp Gly Gly Asp Gly Phe Tyr 115 120
125Ala Met Asp Tyr Trp Gly Gln Gly Thr Leu Val Thr Val Ser
Ser Ala 130 135 140Ser Thr Lys Gly Pro
Ser Val Phe Pro Leu Ala Pro Ser Ser Lys Ser145 150
155 160Thr Ser Gly Gly Thr Ala Ala Leu Gly Cys
Leu Val Lys Asp Tyr Phe 165 170
175Pro Glu Pro Val Thr Val Ser Trp Asn Ser Gly Ala Leu Thr Ser Gly
180 185 190Val His Thr Phe Pro
Ala Val Leu Gln Ser Ser Gly Leu Tyr Ser Leu 195
200 205Ser Ser Val Val Thr Val Pro Ser Ser Ser Leu Gly
Thr Gln Thr Tyr 210 215 220Ile Cys Asn
Val Asn His Lys Pro Ser Asn Thr Lys Val Asp Lys Lys225
230 235 240Val Glu Pro Lys Ser Cys Asp
Lys Thr His Gly Arg Met Lys Gln Leu 245
250 255Glu Asp Lys Val Glu Glu Leu Leu Ser Lys Asn Tyr
His Leu Glu Asn 260 265 270Glu
Val Ala Arg Leu Lys Lys Leu Val Gly Glu Arg Gly Lys Leu Ser 275
280 285Gly Gly Gly Ser Gly Ser Gly Asp Phe
Asp Tyr Glu Lys Met Ala Asn 290 295
300Ala Asn Lys Gly Ala Met Thr Glu Asn Ala Asp Glu Asn Ala Leu Gln305
310 315 320Ser Asp Ala Lys
Gly Lys Leu Asp Ser Val Ala Thr Asp Tyr Gly Ala 325
330 335Ala Ile Asp Gly Phe Ile Gly Asp Val Ser
Gly Leu Ala Asn Gly Asn 340 345
350Gly Ala Thr Gly Asp Phe Ala Gly Ser Asn Ser Gln Met Ala Gln Val
355 360 365Gly Asp Gly Asp Asn Ser Pro
Leu Met Asn Asn Phe Arg Gln Tyr Leu 370 375
380Pro Ser Leu Pro Gln Ser Val Glu Cys Arg Pro Phe Val Phe Ser
Ala385 390 395 400Gly Lys
Pro Tyr Glu Phe Ser Ile Asp Cys Asp Lys Ile Asn Leu Phe
405 410 415Arg Gly Val Phe Ala Phe Leu
Leu Tyr Val Ala Thr Phe Met Tyr Val 420 425
430Phe Ser Thr Phe Ala Asn Ile Leu Arg Asn Lys Glu Ser
435 440 445917171DNAArtificial
SequenceDescription of Artificial Sequence Synthetic polynucleotide
91gaattcaact tctccatact ttggataagg aaatacagac atgaaaaatc tcattgctga
60gttgttattt aagcttgccc aaaaagaaga agagtcgaat gaactgtgtg cgcaggtaga
120agctttggag attatcgtca ctgcaatgct tcgcaatatg gcgcaaaatg accaacagcg
180gttgattgat caggtagagg gggcgctgta cgaggtaaag cccgatgcca gcattcctga
240cgacgatacg gagctgctgc gcgattacgt aaagaagtta ttgaagcatc ctcgtcagta
300aaaagttaat cttttcaaca gctgtcataa agttgtcacg gccgagactt atagtcgctt
360tgtttttatt ttttaatgta tttgtaacta gtacgcaagt tcacgtaaaa agggtatgta
420gaggttgagg tgattttatg aaaaagaata tcgcatttct tcttgcatct atgttcgttt
480tttctattgc tacaaatgcc tatgcatccg atatccagat gacccagtcc ccgagctccc
540tgtccgcctc tgtgggcgat agggtcacca tcacctgccg tgccagtcag gatgtgtcca
600ctgctgtagc ctggtatcaa cagaaaccag gaaaagctcc gaagcttctg atttactcgg
660catccttcct ctactctgga gtcccttctc gcttctctgg tagcggttcc gggacggatt
720tcactctgac catcagcagt ctgcagccgg aagacttcgc aacttattac tgtcagcaat
780cttatactac tcctcccacg ttcggacagg gtaccaaggt ggagatcaaa cgaactgtgg
840ctgcaccatc tgtcttcatc ttcccgccat ctgatgagca gttgaaatct ggaactgcct
900ctgttgtgtg cctgctgaat aacttctatc ccagagaggc caaagtacag tggaaggtgg
960ataacgccct ccaatcgggt aactcccagg agagtgtcac agagcaggac agcaaggaca
1020gcacctacag cctcagcagc accctgacgc tgagcaaagc agactacgag aaacacaaag
1080tctacgcctg cgaagtcacc catcagggcc tgagctcgcc cgtcacaaag agcttcaaca
1140ggggagagtg tggtgccagc tccggtatgg ctgatccgaa ccgtttccgc ggtaaggacc
1200tggcataact cgaggctgat cctctacgcc ggacgcatcg tggccctagt acgcaagttc
1260acgtaaaaag ggtaactaga ggttgaggtg attttatgaa aaagaatatc gcatttcttc
1320ttgcatctat gttcgttttt tctattgcta caaacgcgta cgctgaggtt cagctggtgg
1380agtctggcgg tggcctggtg cagccagggg gctcactccg tttgtcctgt gcagcttctg
1440gcttcaacat taaagacacc tatatacact gggtgcgtca ggccccgggt aagggcctgg
1500aatgggttgc aaggatttat cctacgaatg gttatactag atatgccgat agcgtcaagg
1560gccgtttcac tataagcgca gacacatcca aaaacacagc ctacctacaa atgaacagct
1620taagagctga ggacactgcc gtctattatt gtagccgctg gggaggggac ggcttctatg
1680ctatggacta ctggggtcaa ggaacactag tcaccgtctc ctcggcctcc accaagggcc
1740catcggtctt ccccctggca ccctcctcca agagcacctc tgggggcaca gcggccctgg
1800gctgcctggt caaggactac ttccccgaac cggtgacggt gtcgtggaac tcaggcgccc
1860tgaccagcgg cgtgcacacc ttcccggctg tcctacagtc ctcaggactc tactccctca
1920gcagcgtggt gaccgtgccc tccagcagct tgggcaccca gacctacatc tgcaacgtga
1980atcacaagcc cagcaacacc aaggtcgaca agaaagttga gcccaaatct tgtgacaaaa
2040ctcacggccg catgaaacag ctagaggaca aggtcgaaga gctactctcc aagaactacc
2100acctagagaa tgaagtggca agactcaaaa aacttgtcgg ggagcgcgga aagcttagtg
2160gcggtggctc tggttccggt gattttgatt atgaaaagat ggcaaacgct aataaggggg
2220ctatgaccga aaatgccgat gaaaacgcgc tacagtctga cgctaaaggc aaacttgatt
2280ctgtcgctac tgattacggt gctgctatcg atggtttcat tggtgacgtt tccggccttg
2340ctaatggtaa tggtgctact ggtgattttg ctggctctaa ttcccaaatg gctcaagtcg
2400gtgacggtga taattcacct ttaatgaata atttccgtca atatttacct tccctccctc
2460aatcggttga atgtcgccct tttgtcttta gcgctggtaa accatatgaa ttttctattg
2520attgtgacaa aataaactta ttccgtggtg tctttgcgtt tcttttatat gttgccacct
2580ttatgtatgt attttctacg tttgctaaca tactgcgtaa taaggagtct taatcatgcc
2640agttcttttg gctagcgccg ccctatacct tgtctgcctc cccgcgttgc gtcgcggtgc
2700atggagccgg gccacctcga cctgaatgga agccggcggc acctcgctaa cggattcacc
2760actccaagaa ttggagccaa tcaattcttg cggagaactg tgaatgcgca aaccaaccct
2820tggcagaaca tatccatcgc gtccgccatc tccagcagcc gcacgcggcg catctcgggc
2880agcgttgggt cctggccacg ggtgcgcatg atcgtgctcc tgtcgttgag gacccggcta
2940ggctggcggg gttgccttac tggttagcag aatgaatcac cgatacgcga gcgaacgtga
3000agcgactgct gctgcaaaac gtctgcgacc tgagcaacaa catgaatggt cttcggtttc
3060cgtgtttcgt aaagtctgga aacgcggaag tcagcgccct gcaccattat gttccggatc
3120tgcatcgcag gatgctgctg gctaccctgt ggaacaccta catctgtatt aacgaagcgc
3180tggcattgac cctgagtgat ttttctctgg tcccgccgca tccataccgc cagttgttta
3240ccctcacaac gttccagtaa ccgggcatgt tcatcatcag taacccgtat cgtgagcatc
3300ctctctcgtt tcatcggtat cattaccccc atgaacagaa attccccctt acacggaggc
3360atcaagtgac caaacaggaa aaaaccgccc ttaacatggc ccgctttatc agaagccaga
3420cattaacgct tctggagaaa ctcaacgagc tggacgcgga tgaacaggca gacatctgtg
3480aatcgcttca cgaccacgct gatgagcttt accgcaggat ccggaaattg taaacgttaa
3540tattttgtta aaattcgcgt taaatttttg ttaaatcagc tcatttttta accaataggc
3600cgaaatcggc aaaatccctt ataaatcaaa agaatagacc gagatagggt tgagtgttgt
3660tccagtttgg aacaagagtc cactattaaa gaacgtggac tccaacgtca aagggcgaaa
3720aaccgtctat cagggctatg gcccactacg tgaaccatca ccctaatcaa gttttttggg
3780gtcgaggtgc cgtaaagcac taaatcggaa ccctaaaggg agcccccgat ttagagcttg
3840acggggaaag ccggcgaacg tggcgagaaa ggaagggaag aaagcgaaag gagcgggcgc
3900tagggcgctg gcaagtgtag cggtcacgct gcgcgtaacc accacacccg ccgcgcttaa
3960tgcgccgcta cagggcgcgt ccggatcctg cctcgcgcgt ttcggtgatg acggtgaaaa
4020cctctgacac atgcagctcc cggagacggt cacagcttgt ctgtaagcgg atgccgggag
4080cagacaagcc cgtcagggcg cgtcagcggg tgttggcggg tgtcggggcg cagccatgac
4140ccagtcacgt agcgatagcg gagtgtatac tggcttaact atgcggcatc agagcagatt
4200gtactgagag tgcaccatat gcggtgtgaa ataccgcaca gatgcgtaag gagaaaatac
4260cgcatcaggc gctcttccgc ttcctcgctc actgactcgc tgcgctcggt cgttcggctg
4320cggcgagcgg tatcagctca ctcaaaggcg gtaatacggt tatccacaga atcaggggat
4380aacgcaggaa agaacatgtg agcaaaaggc cagcaaaagg ccaggaaccg taaaaaggcc
4440gcgttgctgg cgtttttcca taggctccgc ccccctgacg agcatcacaa aaatcgacgc
4500tcaagtcaga ggtggcgaaa cccgacagga ctataaagat accaggcgtt tccccctgga
4560agctccctcg tgcgctctcc tgttccgacc ctgccgctta ccggatacct gtccgccttt
4620ctcccttcgg gaagcgtggc gctttctcat agctcacgct gtaggtatct cagttcggtg
4680taggtcgttc gctccaagct gggctgtgtg cacgaacccc ccgttcagcc cgaccgctgc
4740gccttatccg gtaactatcg tcttgagtcc aacccggtaa gacacgactt atcgccactg
4800gcagcagcca ctggtaacag gattagcaga gcgaggtatg taggcggtgc tacagagttc
4860ttgaagtggt ggcctaacta cggctacact agaaggacag tatttggtat ctgcgctctg
4920ctgaagccag ttaccttcgg aaaaagagtt ggtagctctt gatccggcaa acaaaccacc
4980gctggtagcg gtggtttttt tgtttgcaag cagcagatta cgcgcagaaa aaaaggatct
5040caagaagatc ctttgatctt ttctacgggg tctgacgctc agtggaacga aaactcacgt
5100taagggattt tggtcatgag attatcaaaa aggatcttca cctagatcct tttaaattaa
5160aaatgaagtt ttaaatcaat ctaaagtata tatgagtaaa cttggtctga cagttaccaa
5220tgcttaatca gtgaggcacc tatctcagcg atctgtctat ttcgttcatc catagttgcc
5280tgactccccg tcgtgtagat aactacgata cgggagggct taccatctgg ccccagtgct
5340gcaatgatac cgcgagaccc acgctcaccg gctccagatt tatcagcaat aaaccagcca
5400gccggaaggg ccgagcgcag aagtggtcct gcaactttat ccgcctccat ccagtctatt
5460aattgttgcc gggaagctag agtaagtagt tcgccagtta atagtttgcg caacgttgtt
5520gccattgctg caggcatcgt ggtgtcacgc tcgtcgtttg gtatggcttc attcagctcc
5580ggttcccaac gatcaaggcg agttacatga tcccccatgt tgtgcaaaaa agcggttagc
5640tccttcggtc ctccgatcgt tgtcagaagt aagttggccg cagtgttatc actcatggtt
5700atggcagcac tgcataattc tcttactgtc atgccatccg taagatgctt ttctgtgact
5760ggtgagtact caaccaagtc attctgagaa tagtgtatgc ggcgaccgag ttgctcttgc
5820ccggcgtcaa cacgggataa taccgcgcca catagcagaa ctttaaaagt gctcatcatt
5880ggaaaacgtt cttcggggcg aaaactctca aggatcttac cgctgttgag atccagttcg
5940atgtaaccca ctcgtgcacc caactgatct tcagcatctt ttactttcac cagcgtttct
6000gggtgagcaa aaacaggaag gcaaaatgcc gcaaaaaagg gaataagggc gacacggaaa
6060tgttgaatac tcatactctt cctttttcaa tattattgaa gcatttatca gggttattgt
6120ctcatgagcg gatacatatt tgaatgtatt tagaaaaata aacaaatagg ggttccgcgc
6180acatttcccc gaaaagtgcc acctgacgtc taagaaacca ttattatcat gacattaacc
6240tataaaaata ggcgtatcac gaggcccttt cgtcttcaat acaggtagac ctttcgtaga
6300gatgtacagt gaaatccccg aaattataca catgactgaa ggaagggagc tcgtcattcc
6360ctgccgggtt acgtcaccta acatcactgt tactttaaaa aagtttccac ttgacacttt
6420gatccctgat ggaaaacgca taatctggga cagtagaaag ggcttcatca tatcaaatgc
6480aacgtacaaa gaaatagggc ttctgacctg tgaagcaaca gtcaatgggc atttgtataa
6540gacaaactat ctcacacatc gacaaaccaa tacaatacag gtagaccttt cgtagagatg
6600tacagtgaaa tccccgaaat tatacacatg actgaaggaa gggagctcgt cattccctgc
6660cgggttacgt cacctaacat cactgttact ttaaaaaagt ttccacttga cactttgatc
6720cctgatggaa aacgcataat ctgggacagt agaaagggct tcatcatatc aaatgcaacg
6780tacaaagaaa tagggcttct gacctgtgaa gcaacagtca atgggcattt gtataagaca
6840aactatctca cacatcgaca aaccaataca atctacaggt agacctttcg tagagatgta
6900cagtgaaatc cccgaaatta tacacatgac tgaaggaagg gagctcgtca ttccctgccg
6960ggttacgtca cctaacatca ctgttacttt aaaaaagttt ccacttgaca ctttgatccc
7020tgatggaaaa cgcataatct gggacagtag aaagggcttc atcatatcaa atgcaacgta
7080caaagaaata gggcttctga cctgtgaagc aacagtcaat gggcatttgt ataagacaaa
7140ctatctcaca catcgacaaa ccaatacaat c
717192255PRTArtificial SequenceDescription of Artificial Sequence
Synthetic polypeptide 92Met Lys Lys Asn Ile Ala Phe Leu Leu Ala Ser
Met Phe Val Phe Ser1 5 10
15Ile Ala Thr Asn Ala Tyr Ala Asp Ile Gln Met Thr Gln Ser Pro Ser
20 25 30Ser Leu Ser Ala Ser Val Gly
Asp Arg Val Thr Ile Thr Cys Arg Ala 35 40
45Ser Gln Asp Val Ser Thr Ala Val Ala Trp Tyr Gln Gln Lys Pro
Gly 50 55 60Lys Ala Pro Lys Leu Leu
Ile Tyr Ser Ala Ser Phe Leu Tyr Ser Gly65 70
75 80Val Pro Ser Arg Phe Ser Gly Ser Gly Ser Gly
Thr Asp Phe Thr Leu 85 90
95Thr Ile Ser Ser Leu Gln Pro Glu Asp Phe Ala Thr Tyr Tyr Cys Gln
100 105 110Gln His Tyr Thr Thr Pro
Pro Thr Phe Gly Gln Gly Thr Lys Val Glu 115 120
125Ile Lys Arg Thr Val Ala Ala Pro Ser Val Phe Ile Phe Pro
Pro Ser 130 135 140Asp Glu Gln Leu Lys
Ser Gly Thr Ala Ser Val Val Cys Leu Leu Asn145 150
155 160Asn Phe Tyr Pro Arg Glu Ala Lys Val Gln
Trp Lys Val Asp Asn Ala 165 170
175Leu Gln Ser Gly Asn Ser Gln Glu Ser Val Thr Glu Gln Asp Ser Lys
180 185 190Asp Ser Thr Tyr Ser
Leu Ser Ser Thr Leu Thr Leu Ser Lys Ala Asp 195
200 205Tyr Glu Lys His Lys Val Tyr Ala Cys Glu Val Thr
His Gln Gly Leu 210 215 220Ser Ser Pro
Val Thr Lys Ser Phe Asn Arg Gly Glu Cys Gly Ala Ser225
230 235 240Ser Gly Met Ala Asp Pro Asn
Arg Phe Arg Gly Lys Asp Leu Ala 245 250
25593408PRTArtificial SequenceDescription of Artificial
Sequence Synthetic polypeptide 93Met Lys Lys Asn Ile Ala Phe Leu Leu
Ala Ser Met Phe Val Phe Ser1 5 10
15Ile Ala Thr Asn Ala Tyr Ala Glu Val Gln Leu Val Glu Ser Gly
Gly 20 25 30Gly Leu Val Gln
Pro Gly Gly Ser Leu Arg Leu Ser Cys Ala Ala Ser 35
40 45Gly Phe Thr Ile Ser Gly Ser Trp Ile His Trp Val
Arg Gln Ala Pro 50 55 60Gly Lys Gly
Leu Glu Trp Val Ala Trp Ile Ala Pro Tyr Ser Gly Ala65 70
75 80Thr Asp Tyr Ala Asp Ser Val Lys
Gly Arg Phe Thr Ile Ser Ala Asp 85 90
95Thr Ser Lys Asn Thr Ala Tyr Leu Gln Met Asn Ser Leu Arg
Ala Glu 100 105 110Asp Thr Ala
Val Tyr Tyr Cys Ala Arg Glu Gly Gly Leu Tyr Trp Val 115
120 125Phe Asp Tyr Trp Gly Gln Gly Thr Leu Val Thr
Val Ser Ser Ala Ser 130 135 140Thr Lys
Gly Pro Ser Val Phe Pro Leu Ala Pro Ser Ser Lys Ser Thr145
150 155 160Ser Gly Gly Thr Ala Ala Leu
Gly Cys Leu Val Lys Asp Tyr Phe Pro 165
170 175Glu Pro Val Thr Val Ser Trp Asn Ser Gly Ala Leu
Thr Ser Gly Val 180 185 190His
Thr Phe Pro Ala Val Leu Gln Ser Ser Gly Leu Tyr Ser Leu Ser 195
200 205Ser Val Val Thr Val Pro Ser Ser Ser
Leu Gly Thr Gln Thr Tyr Ile 210 215
220Cys Asn Val Asn His Lys Pro Ser Asn Thr Lys Val Asp Lys Lys Val225
230 235 240Glu Pro Lys Ser
Cys Asp Lys Thr His Leu Ser Gly Gly Gly Ser Gly 245
250 255Ser Gly Asp Phe Asp Tyr Glu Lys Met Ala
Asn Ala Asn Lys Gly Ala 260 265
270Met Thr Glu Asn Ala Asp Glu Asn Ala Leu Gln Ser Asp Ala Lys Gly
275 280 285Lys Leu Asp Ser Val Ala Thr
Asp Tyr Gly Ala Ala Ile Asp Gly Phe 290 295
300Ile Gly Asp Val Ser Gly Leu Ala Asn Gly Asn Gly Ala Thr Gly
Asp305 310 315 320Phe Ala
Gly Ser Asn Ser Gln Met Ala Gln Val Gly Asp Gly Asp Asn
325 330 335Ser Pro Leu Met Asn Asn Phe
Arg Gln Tyr Leu Pro Ser Leu Pro Gln 340 345
350Ser Val Glu Cys Arg Pro Phe Val Phe Ser Ala Gly Lys Pro
Tyr Glu 355 360 365Phe Ser Ile Asp
Cys Asp Lys Ile Asn Leu Phe Arg Gly Val Phe Ala 370
375 380Phe Leu Leu Tyr Val Ala Thr Phe Met Tyr Val Phe
Ser Thr Phe Ala385 390 395
400Asn Ile Leu Arg Asn Lys Glu Ser 405942082DNAArtificial
SequenceDescription of Artificial Sequence Synthetic polynucleotide
94atgaaaaaga atatcgcatt tcttcttgca tctatgttcg ttttttctat tgctacaaat
60gcctatgcag atatccagat gacccagtcc ccgagctccc tgtccgcctc tgtgggcgat
120agggtcacca tcacctgccg tgccagtcag gatgtgtcca ctgctgtagc ctggtatcaa
180cagaaaccag gaaaagctcc gaagcttctg atttactcgg catccttcct ctactctgga
240gtcccttctc gcttctctgg tagcggttcc gggacggatt tcactctgac catcagcagt
300ctgcagccgg aagacttcgc aacttattac tgtcagcaac attatactac tcctcccacg
360ttcggacagg gtaccaaggt ggagatcaaa cgaactgtgg ctgcaccatc tgtcttcatc
420ttcccgccat ctgatgagca gttgaaatct ggaactgcct ctgttgtgtg cctgctgaat
480aacttctatc ccagagaggc caaagtacag tggaaggtgg ataacgccct ccaatcgggt
540aactcccagg agagtgtcac agagcaggac agcaaggaca gcacctacag cctcagcagc
600accctgacgc tgagcaaagc agactacgag aaacacaaag tctacgcctg cgaagtcacc
660catcagggcc tgagctcgcc cgtcacaaag agcttcaaca ggggagagtg tggtgccagc
720tccggtatgg ctgatccgaa ccgtttccgc ggtaaggacc tggcataact cgaggctgat
780cctctacgcc ggacgcatcg tggccctagt acgcaagttc acgtaaaaag ggtaactaga
840ggttgaggtg attttatgaa aaagaatatc gcatttcttc ttgcatctat gttcgttttt
900tctattgcta caaacgcgta cgctgaggtt cagctggtgg agtctggcgg tggcctggtg
960cagccagggg gctcactccg tttgtcctgt gcagcttctg gcttcaccat tagtggttct
1020tggatacact gggtgcgtca ggccccgggt aagggcctgg aatgggttgc ttggattgct
1080ccttatagcg gcgctactga ctatgccgat agcgtcaagg gccgtttcac tataagcgca
1140gacacatcca aaaacacagc ctacctacaa atgaacagct taagagctga ggacactgcc
1200gtctattatt gtgcaagaga ggggggcttg tactgggtgt tcgactactg gggtcaagga
1260acactagtca ccgtctcctc ggcctccacc aagggcccat cggtcttccc cctggcaccc
1320tcctccaaga gcacctctgg gggcacagcg gccctgggct gcctggtcaa ggactacttc
1380cccgaaccgg tgacggtgtc gtggaactca ggcgccctga ccagcggcgt gcacaccttc
1440ccggctgtcc tacagtcctc aggactctac tccctcagca gcgtggtgac cgtgccctcc
1500agcagcttgg gcacccagac ctacatctgc aacgtgaatc acaagcccag caacaccaag
1560gtcgacaaga aagttgagcc caaatcttgt gacaaaactc acctcagtgg cggtggctct
1620ggttccggtg attttgatta tgaaaagatg gcaaacgcta ataagggggc tatgaccgaa
1680aatgccgatg aaaacgcgct acagtctgac gctaaaggca aacttgattc tgtcgctact
1740gattacggtg ctgctatcga tggtttcatt ggtgacgttt ccggccttgc taatggtaat
1800ggtgctactg gtgattttgc tggctctaat tcccaaatgg ctcaagtcgg tgacggtgat
1860aattcacctt taatgaataa tttccgtcaa tatttacctt ccctccctca atcggttgaa
1920tgtcgccctt ttgtctttag cgctggtaaa ccatatgaat tttctattga ttgtgacaaa
1980ataaacttat tccgtggtgt ctttgcgttt cttttatatg ttgccacctt tatgtatgta
2040ttttctacgt ttgctaacat actgcgtaat aaggagtctt aa
208295255PRTArtificial SequenceDescription of Artificial Sequence
Synthetic polypeptide 95Met Lys Lys Asn Ile Ala Phe Leu Leu Ala Ser
Met Phe Val Phe Ser1 5 10
15Ile Ala Thr Asn Ala Tyr Ala Asp Ile Gln Met Thr Gln Ser Pro Ser
20 25 30Ser Leu Ser Ala Ser Val Gly
Asp Arg Val Thr Ile Thr Cys Arg Ala 35 40
45Ser Gln Asp Val Ser Thr Ala Val Ala Trp Tyr Gln Gln Lys Pro
Gly 50 55 60Lys Ala Pro Lys Leu Leu
Ile Tyr Ser Ala Ser Phe Leu Tyr Ser Gly65 70
75 80Val Pro Ser Arg Phe Ser Gly Ser Gly Ser Gly
Thr Asp Phe Thr Leu 85 90
95Thr Ile Ser Ser Leu Gln Pro Glu Asp Phe Ala Thr Tyr Tyr Cys Gln
100 105 110Gln His Tyr Thr Thr Pro
Pro Thr Phe Gly Gln Gly Thr Lys Val Glu 115 120
125Ile Lys Arg Thr Val Ala Ala Pro Ser Val Phe Ile Phe Pro
Pro Ser 130 135 140Asp Glu Gln Leu Lys
Ser Gly Thr Ala Ser Val Val Cys Leu Leu Asn145 150
155 160Asn Phe Tyr Pro Arg Glu Ala Lys Val Gln
Trp Lys Val Asp Asn Ala 165 170
175Leu Gln Ser Gly Asn Ser Gln Glu Ser Val Thr Glu Gln Asp Ser Lys
180 185 190Asp Ser Thr Tyr Ser
Leu Ser Ser Thr Leu Thr Leu Ser Lys Ala Asp 195
200 205Tyr Glu Lys His Lys Val Tyr Ala Cys Glu Val Thr
His Gln Gly Leu 210 215 220Ser Ser Pro
Val Thr Lys Ser Phe Asn Arg Gly Glu Cys Gly Ala Ser225
230 235 240Ser Gly Met Ala Asp Pro Asn
Arg Phe Arg Gly Lys Asp Leu Ala 245 250
25596405PRTArtificial SequenceDescription of Artificial
Sequence Synthetic polypeptide 96Met Lys Lys Asn Ile Ala Phe Leu Leu
Ala Ser Met Phe Val Phe Ser1 5 10
15Ile Ala Thr Asn Ala Tyr Ala Glu Val Gln Leu Val Glu Ser Gly
Gly 20 25 30Gly Leu Val Gln
Pro Gly Gly Ser Leu Arg Leu Ser Cys Ala Ala Ser 35
40 45Gly Phe Thr Ile Ser Asn Tyr Gly Ile His Trp Val
Arg Gln Ala Pro 50 55 60Gly Lys Gly
Leu Glu Trp Val Gly Arg Ile Ser Pro Ser Asn Gly Ser65 70
75 80Thr Tyr Tyr Ala Asp Ser Val Lys
Gly Arg Phe Thr Ile Ser Ala Asp 85 90
95Thr Ser Lys Asn Thr Ala Tyr Leu Gln Met Asn Ser Leu Arg
Ala Glu 100 105 110Asp Thr Ala
Val Tyr Tyr Cys Ala Lys Cys Ser Val Arg Phe Ala Tyr 115
120 125Trp Gly Gln Gly Thr Leu Val Thr Val Ser Ser
Ala Ser Thr Lys Gly 130 135 140Pro Ser
Val Phe Pro Leu Ala Pro Ser Ser Lys Ser Thr Ser Gly Gly145
150 155 160Thr Ala Ala Leu Gly Cys Leu
Val Lys Asp Tyr Phe Pro Glu Pro Val 165
170 175Thr Val Ser Trp Asn Ser Gly Ala Leu Thr Ser Gly
Val His Thr Phe 180 185 190Pro
Ala Val Leu Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val 195
200 205Thr Val Pro Ser Ser Ser Leu Gly Thr
Gln Thr Tyr Ile Cys Asn Val 210 215
220Asn His Lys Pro Ser Asn Thr Lys Val Asp Lys Lys Val Glu Pro Lys225
230 235 240Ser Cys Asp Lys
Thr His Leu Ser Gly Gly Gly Ser Gly Ser Gly Asp 245
250 255Phe Asp Tyr Glu Lys Met Ala Asn Ala Asn
Lys Gly Ala Met Thr Glu 260 265
270Asn Ala Asp Glu Asn Ala Leu Gln Ser Asp Ala Lys Gly Lys Leu Asp
275 280 285Ser Val Ala Thr Asp Tyr Gly
Ala Ala Ile Asp Gly Phe Ile Gly Asp 290 295
300Val Ser Gly Leu Ala Asn Gly Asn Gly Ala Thr Gly Asp Phe Ala
Gly305 310 315 320Ser Asn
Ser Gln Met Ala Gln Val Gly Asp Gly Asp Asn Ser Pro Leu
325 330 335Met Asn Asn Phe Arg Gln Tyr
Leu Pro Ser Leu Pro Gln Ser Val Glu 340 345
350Cys Arg Pro Phe Val Phe Ser Ala Gly Lys Pro Tyr Glu Phe
Ser Ile 355 360 365Asp Cys Asp Lys
Ile Asn Leu Phe Arg Gly Val Phe Ala Phe Leu Leu 370
375 380Tyr Val Ala Thr Phe Met Tyr Val Phe Ser Thr Phe
Ala Asn Ile Leu385 390 395
400Arg Asn Lys Glu Ser 405972073DNAArtificial
SequenceDescription of Artificial Sequence Synthetic polynucleotide
97atgaaaaaga atatcgcatt tcttcttgca tctatgttcg ttttttctat tgctacaaat
60gcctatgcag atatccagat gacccagtcc ccgagctccc tgtccgcctc tgtgggcgat
120agggtcacca tcacctgccg tgccagtcag gatgtgtcca ctgctgtagc ctggtatcaa
180cagaaaccag gaaaagctcc gaagcttctg atttactcgg catccttcct ctactctgga
240gtcccttctc gcttctctgg tagcggttcc gggacggatt tcactctgac catcagcagt
300ctgcagccgg aagacttcgc aacttattac tgtcagcaac attatactac tcctcccacg
360ttcggacagg gtaccaaggt ggagatcaaa cgaactgtgg ctgcaccatc tgtcttcatc
420ttcccgccat ctgatgagca gttgaaatct ggaactgcct ctgttgtgtg cctgctgaat
480aacttctatc ccagagaggc caaagtacag tggaaggtgg ataacgccct ccaatcgggt
540aactcccagg agagtgtcac agagcaggac agcaaggaca gcacctacag cctcagcagc
600accctgacgc tgagcaaagc agactacgag aaacacaaag tctacgcctg cgaagtcacc
660catcagggcc tgagctcgcc cgtcacaaag agcttcaaca ggggagagtg tggtgccagc
720tccggtatgg ctgatccgaa ccgtttccgc ggtaaggacc tggcataact cgaggctgat
780cctctacgcc ggacgcatcg tggccctagt acgcaagttc acgtaaaaag ggtaactaga
840ggttgaggtg attttatgaa aaagaatatc gcatttcttc ttgcatctat gttcgttttt
900tctattgcta caaacgcgta cgctgaggtt cagctggtgg agtctggcgg tggcctggtg
960cagccagggg gctcactccg tttgtcctgt gcagcttctg gcttcaccat tagtaattat
1020gggatacact gggtgcgtca ggccccgggt aagggcctgg aatgggttgg taggatttct
1080ccttctaacg gctctactta ctatgccgat agcgtcaagg gccgtttcac tataagcgca
1140gacacatcca aaaacacagc ctacctacaa atgaacagct taagagctga ggacactgcc
1200gtctattatt gtgcaaaatg ctcggtcagg ttcgcttact ggggtcaagg aacactagtc
1260accgtctcct cggcctccac caagggccca tcggtcttcc ccctggcacc ctcctccaag
1320agcacctctg ggggcacagc ggccctgggc tgcctggtca aggactactt ccccgaaccg
1380gtgacggtgt cgtggaactc aggcgccctg accagcggcg tgcacacctt cccggctgtc
1440ctacagtcct caggactcta ctccctcagc agcgtggtga ccgtgccctc cagcagcttg
1500ggcacccaga cctacatctg caacgtgaat cacaagccca gcaacaccaa ggtcgacaag
1560aaagttgagc ccaaatcttg tgacaaaact cacctcagtg gcggtggctc tggttccggt
1620gattttgatt atgaaaagat ggcaaacgct aataaggggg ctatgaccga aaatgccgat
1680gaaaacgcgc tacagtctga cgctaaaggc aaacttgatt ctgtcgctac tgattacggt
1740gctgctatcg atggtttcat tggtgacgtt tccggccttg ctaatggtaa tggtgctact
1800ggtgattttg ctggctctaa ttcccaaatg gctcaagtcg gtgacggtga taattcacct
1860ttaatgaata atttccgtca atatttacct tccctccctc aatcggttga atgtcgccct
1920tttgtcttta gcgctggtaa accatatgaa ttttctattg attgtgacaa aataaactta
1980ttccgtggtg tctttgcgtt tcttttatat gttgccacct ttatgtatgt attttctacg
2040tttgctaaca tactgcgtaa taaggagtct taa
207398256PRTArtificial SequenceDescription of Artificial Sequence
Synthetic polypeptide 98Met Lys Lys Asn Ile Ala Phe Leu Leu Ala Ser
Met Phe Val Phe Ser1 5 10
15Ile Ala Thr Asn Ala Tyr Ala Ser Asp Ile Gln Met Thr Gln Ser Pro
20 25 30Ser Ser Leu Ser Ala Ser Val
Gly Asp Arg Val Thr Ile Thr Cys Arg 35 40
45Ala Ser Gln Asp Val Ser Thr Ala Val Ala Trp Tyr Gln Gln Lys
Pro 50 55 60Gly Lys Ala Pro Lys Leu
Leu Ile Tyr Ser Ala Ser Phe Leu Tyr Ser65 70
75 80Gly Val Pro Ser Arg Phe Ser Gly Ser Gly Ser
Gly Thr Asp Phe Thr 85 90
95Leu Thr Ile Ser Ser Leu Gln Pro Glu Asp Phe Ala Thr Tyr Tyr Cys
100 105 110Gln Gln Ser Tyr Thr Thr
Pro Pro Thr Phe Gly Gln Gly Thr Lys Val 115 120
125Glu Ile Lys Arg Thr Val Ala Ala Pro Ser Val Phe Ile Phe
Pro Pro 130 135 140Ser Asp Glu Gln Leu
Lys Ser Gly Thr Ala Ser Val Val Cys Leu Leu145 150
155 160Asn Asn Phe Tyr Pro Arg Glu Ala Lys Val
Gln Trp Lys Val Asp Asn 165 170
175Ala Leu Gln Ser Gly Asn Ser Gln Glu Ser Val Thr Glu Gln Asp Ser
180 185 190Lys Asp Ser Thr Tyr
Ser Leu Ser Ser Thr Leu Thr Leu Ser Lys Ala 195
200 205Asp Tyr Glu Lys His Lys Val Tyr Ala Cys Glu Val
Thr His Gln Gly 210 215 220Leu Ser Ser
Pro Val Thr Lys Ser Phe Asn Arg Gly Glu Cys Gly Ala225
230 235 240Ser Ser Gly Met Ala Asp Pro
Asn Arg Phe Arg Gly Lys Asp Leu Ala 245
250 25599444PRTArtificial SequenceDescription of
Artificial Sequence Synthetic polypeptide 99Met Lys Lys Asn Ile Ala
Phe Leu Leu Ala Ser Met Phe Val Phe Ser1 5
10 15Ile Ala Thr Asn Ala Tyr Ala Glu Val Gln Leu Val
Glu Ser Gly Gly 20 25 30Gly
Leu Val Gln Pro Gly Gly Ser Leu Arg Leu Ser Cys Ala Ala Ser 35
40 45Gly Phe Thr Ile Ser Gly Ser Asp Ile
His Trp Val Arg Gln Ala Pro 50 55
60Gly Lys Gly Leu Glu Trp Val Gly Arg Ile Ser Pro Tyr Gly Gly Asn65
70 75 80Thr Asn Tyr Ala Asp
Ser Val Lys Gly Arg Phe Thr Ile Ser Ala Asp 85
90 95Thr Ser Lys Asn Thr Ala Tyr Leu Gln Met Asn
Ser Leu Arg Ala Glu 100 105
110Asp Thr Ala Val Tyr Tyr Cys Ala Arg Val Gly Gly Leu Lys Leu Leu
115 120 125Phe Asp Tyr Trp Gly Gln Gly
Thr Leu Val Thr Val Ser Ser Ala Ser 130 135
140Thr Lys Gly Pro Ser Val Phe Pro Leu Ala Pro Ser Ser Lys Ser
Thr145 150 155 160Ser Gly
Gly Thr Ala Ala Leu Gly Cys Leu Val Lys Asp Tyr Phe Pro
165 170 175Glu Pro Val Thr Val Ser Trp
Asn Ser Gly Ala Leu Thr Ser Gly Val 180 185
190His Thr Phe Pro Ala Val Leu Gln Ser Ser Gly Leu Tyr Ser
Leu Ser 195 200 205Ser Val Val Thr
Val Pro Ser Ser Ser Leu Gly Thr Gln Thr Tyr Ile 210
215 220Cys Asn Val Asn His Lys Pro Ser Asn Thr Lys Val
Asp Lys Lys Val225 230 235
240Glu Pro Lys Ser Cys Asp Lys Thr His Gly Arg Met Lys Gln Leu Glu
245 250 255Asp Lys Val Glu Glu
Leu Leu Ser Lys Asn Tyr His Leu Glu Asn Glu 260
265 270Val Ala Arg Leu Lys Lys Leu Val Gly Glu Arg Gly
Lys Leu Ser Gly 275 280 285Gly Gly
Ser Gly Ser Gly Asp Phe Asp Tyr Glu Lys Met Ala Asn Ala 290
295 300Asn Lys Gly Ala Met Thr Glu Asn Ala Asp Glu
Asn Ala Leu Gln Ser305 310 315
320Asp Ala Lys Gly Lys Leu Asp Ser Val Ala Thr Asp Tyr Gly Ala Ala
325 330 335Ile Asp Gly Phe
Ile Gly Asp Val Ser Gly Leu Ala Asn Gly Asn Gly 340
345 350Ala Thr Gly Asp Phe Ala Gly Ser Asn Ser Gln
Met Ala Gln Val Gly 355 360 365Asp
Gly Asp Asn Ser Pro Leu Met Asn Asn Phe Arg Gln Tyr Leu Pro 370
375 380Ser Leu Pro Gln Ser Val Glu Cys Arg Pro
Phe Val Phe Ser Ala Gly385 390 395
400Lys Pro Tyr Glu Phe Ser Ile Asp Cys Asp Lys Ile Asn Leu Phe
Arg 405 410 415Gly Val Phe
Ala Phe Leu Leu Tyr Val Ala Thr Phe Met Tyr Val Phe 420
425 430Ser Thr Phe Ala Asn Ile Leu Arg Asn Lys
Glu Ser 435 4401002193DNAArtificial
SequenceDescription of Artificial Sequence Synthetic polynucleotide
100atgaaaaaga atatcgcatt tcttcttgca tctatgttcg ttttttctat tgctacaaat
60gcctatgcat ccgatatcca gatgacccag tccccgagct ccctgtccgc ctctgtgggc
120gatagggtca ccatcacctg ccgtgccagt caggatgtgt ccactgctgt agcctggtat
180caacagaaac caggaaaagc tccgaagctt ctgatttact cggcatcctt cctctactct
240ggagtccctt ctcgcttctc tggtagcggt tccgggacgg atttcactct gaccatcagc
300agtctgcagc cggaagactt cgcaacttat tactgtcagc aatcttatac tactcctccc
360acgttcggac agggtaccaa ggtggagatc aaacgaactg tggctgcacc atctgtcttc
420atcttcccgc catctgatga gcagttgaaa tctggaactg cctctgttgt gtgcctgctg
480aataacttct atcccagaga ggccaaagta cagtggaagg tggataacgc cctccaatcg
540ggtaactccc aggagagtgt cacagagcag gacagcaagg acagcaccta cagcctcagc
600agcaccctga cgctgagcaa agcagactac gagaaacaca aagtctacgc ctgcgaagtc
660acccatcagg gcctgagctc gcccgtcaca aagagcttca acaggggaga gtgtggtgcc
720agctccggta tggctgatcc gaaccgtttc cgcggtaagg acctggcata actcgaggct
780gatcctctac gccggacgca tcgtggccct agtacgcaag ttcacgtaaa aagggtaact
840agaggttgag gtgattttat gaaaaagaat atcgcatttc ttcttgcatc tatgttcgtt
900ttttctattg ctacaaacgc gtacgctgag gttcagctgg tggagtctgg cggtggcctg
960gtgcagccag ggggctcact ccgtttgtcc tgtgcagctt ctggcttcac cattagtggt
1020tctgatatac actgggtgcg tcaggccccg ggtaagggcc tggaatgggt tggtaggatt
1080tctccttatg gcggcaatac taactatgcc gatagcgtca agggccgttt cactataagc
1140gcagacacat ccaaaaacac agcctaccta caaatgaaca gcttaagagc tgaggacact
1200gccgtctatt attgtgcaag agtcggcggc ctcaagttgc tgttcgacta ctggggtcaa
1260ggaacactag tcaccgtctc ctcggcctcc accaagggcc catcggtctt ccccctggca
1320ccctcctcca agagcacctc tgggggcaca gcggccctgg gctgcctggt caaggactac
1380ttccccgaac cggtgacggt gtcgtggaac tcaggcgccc tgaccagcgg cgtgcacacc
1440ttcccggctg tcctacagtc ctcaggactc tactccctca gcagcgtggt gaccgtgccc
1500tccagcagct tgggcaccca gacctacatc tgcaacgtga atcacaagcc cagcaacacc
1560aaggtcgaca agaaagttga gcccaaatct tgtgacaaaa ctcacggccg catgaaacag
1620ctagaggaca aggtcgaaga gctactctcc aagaactacc acctagagaa tgaagtggca
1680agactcaaaa aacttgtcgg ggagcgcgga aagcttagtg gcggtggctc tggttccggt
1740gattttgatt atgaaaagat ggcaaacgct aataaggggg ctatgaccga aaatgccgat
1800gaaaacgcgc tacagtctga cgctaaaggc aaacttgatt ctgtcgctac tgattacggt
1860gctgctatcg atggtttcat tggtgacgtt tccggccttg ctaatggtaa tggtgctact
1920ggtgattttg ctggctctaa ttcccaaatg gctcaagtcg gtgacggtga taattcacct
1980ttaatgaata atttccgtca atatttacct tccctccctc aatcggttga atgtcgccct
2040tttgtcttta gcgctggtaa accatatgaa ttttctattg attgtgacaa aataaactta
2100ttccgtggtg tctttgcgtt tcttttatat gttgccacct ttatgtatgt attttctacg
2160tttgctaaca tactgcgtaa taaggagtct taa
2193101256PRTArtificial SequenceDescription of Artificial Sequence
Synthetic polypeptide 101Met Lys Lys Asn Ile Ala Phe Leu Leu Ala Ser
Met Phe Val Phe Ser1 5 10
15Ile Ala Thr Asn Ala Tyr Ala Ser Asp Ile Gln Met Thr Gln Ser Pro
20 25 30Ser Ser Leu Ser Ala Ser Val
Gly Asp Arg Val Thr Ile Thr Cys Arg 35 40
45Ala Ser Gln Asp Val Ser Thr Ala Val Ala Trp Tyr Gln Gln Lys
Pro 50 55 60Gly Lys Ala Pro Lys Leu
Leu Ile Tyr Ser Ala Ser Phe Leu Tyr Ser65 70
75 80Gly Val Pro Ser Arg Phe Ser Gly Ser Gly Ser
Gly Thr Asp Phe Thr 85 90
95Leu Thr Ile Ser Ser Leu Gln Pro Glu Asp Phe Ala Thr Tyr Tyr Cys
100 105 110Gln Gln Ser Tyr Thr Thr
Pro Pro Thr Phe Gly Gln Gly Thr Lys Val 115 120
125Glu Ile Lys Arg Thr Val Ala Ala Pro Ser Val Phe Ile Phe
Pro Pro 130 135 140Ser Asp Glu Gln Leu
Lys Ser Gly Thr Ala Ser Val Val Cys Leu Leu145 150
155 160Asn Asn Phe Tyr Pro Arg Glu Ala Lys Val
Gln Trp Lys Val Asp Asn 165 170
175Ala Leu Gln Ser Gly Asn Ser Gln Glu Ser Val Thr Glu Gln Asp Ser
180 185 190Lys Asp Ser Thr Tyr
Ser Leu Ser Ser Thr Leu Thr Leu Ser Lys Ala 195
200 205Asp Tyr Glu Lys His Lys Val Tyr Ala Cys Glu Val
Thr His Gln Gly 210 215 220Leu Ser Ser
Pro Val Thr Lys Ser Phe Asn Arg Gly Glu Cys Gly Ala225
230 235 240Ser Ser Gly Met Ala Asp Pro
Asn Arg Phe Arg Gly Lys Asp Leu Ala 245
250 255102445PRTArtificial SequenceDescription of
Artificial Sequence Synthetic polypeptide 102Met Lys Lys Asn Ile Ala
Phe Leu Leu Ala Ser Met Phe Val Phe Ser1 5
10 15Ile Ala Thr Asn Ala Tyr Ala Glu Val Gln Leu Val
Glu Ser Gly Gly 20 25 30Gly
Leu Val Gln Pro Gly Gly Ser Leu Arg Leu Ser Cys Ala Ala Ser 35
40 45Gly Phe Thr Ile Thr Asn Ser Asp Ile
His Trp Val Arg Gln Ala Pro 50 55
60Gly Lys Gly Leu Glu Trp Val Ala Thr Ile Tyr Pro Tyr Gly Gly Tyr65
70 75 80Thr Tyr Tyr Ala Asp
Ser Val Lys Gly Arg Phe Thr Ile Ser Ala Asp 85
90 95Thr Ser Lys Asn Thr Ala Tyr Leu Gln Met Asn
Ser Leu Arg Ala Glu 100 105
110Asp Thr Ala Val Tyr Tyr Cys Ala Arg Gly Gly Gly Met Asp Gly Tyr
115 120 125Val Met Asp Tyr Trp Gly Gln
Gly Thr Leu Val Thr Val Ser Ser Ala 130 135
140Ser Thr Lys Gly Pro Ser Val Phe Pro Leu Ala Pro Ser Ser Lys
Ser145 150 155 160Thr Ser
Gly Gly Thr Ala Ala Leu Gly Cys Leu Val Lys Asp Tyr Phe
165 170 175Pro Glu Pro Val Thr Val Ser
Trp Asn Ser Gly Ala Leu Thr Ser Gly 180 185
190Val His Thr Phe Pro Ala Val Leu Gln Ser Ser Gly Leu Tyr
Ser Leu 195 200 205Ser Ser Val Val
Thr Val Pro Ser Ser Ser Leu Gly Thr Gln Thr Tyr 210
215 220Ile Cys Asn Val Asn His Lys Pro Ser Asn Thr Lys
Val Asp Lys Lys225 230 235
240Val Glu Pro Lys Ser Cys Asp Lys Thr His Gly Arg Met Lys Gln Leu
245 250 255Glu Asp Lys Val Glu
Glu Leu Leu Ser Lys Asn Tyr His Leu Glu Asn 260
265 270Glu Val Ala Arg Leu Lys Lys Leu Val Gly Glu Arg
Gly Lys Leu Ser 275 280 285Gly Gly
Gly Ser Gly Ser Gly Asp Phe Asp Tyr Glu Lys Met Ala Asn 290
295 300Ala Asn Lys Gly Ala Met Thr Glu Asn Ala Asp
Glu Asn Ala Leu Gln305 310 315
320Ser Asp Ala Lys Gly Lys Leu Asp Ser Val Ala Thr Asp Tyr Gly Ala
325 330 335Ala Ile Asp Gly
Phe Ile Gly Asp Val Ser Gly Leu Ala Asn Gly Asn 340
345 350Gly Ala Thr Gly Asp Phe Ala Gly Ser Asn Ser
Gln Met Ala Gln Val 355 360 365Gly
Asp Gly Asp Asn Ser Pro Leu Met Asn Asn Phe Arg Gln Tyr Leu 370
375 380Pro Ser Leu Pro Gln Ser Val Glu Cys Arg
Pro Phe Val Phe Ser Ala385 390 395
400Gly Lys Pro Tyr Glu Phe Ser Ile Asp Cys Asp Lys Ile Asn Leu
Phe 405 410 415Arg Gly Val
Phe Ala Phe Leu Leu Tyr Val Ala Thr Phe Met Tyr Val 420
425 430Phe Ser Thr Phe Ala Asn Ile Leu Arg Asn
Lys Glu Ser 435 440
4451032196DNAArtificial SequenceDescription of Artificial Sequence
Synthetic polynucleotide 103atgaaaaaga atatcgcatt tcttcttgca
tctatgttcg ttttttctat tgctacaaat 60gcctatgcat ccgatatcca gatgacccag
tccccgagct ccctgtccgc ctctgtgggc 120gatagggtca ccatcacctg ccgtgccagt
caggatgtgt ccactgctgt agcctggtat 180caacagaaac caggaaaagc tccgaagctt
ctgatttact cggcatcctt cctctactct 240ggagtccctt ctcgcttctc tggtagcggt
tccgggacgg atttcactct gaccatcagc 300agtctgcagc cggaagactt cgcaacttat
tactgtcagc aatcttatac tactcctccc 360acgttcggac agggtaccaa ggtggagatc
aaacgaactg tggctgcacc atctgtcttc 420atcttcccgc catctgatga gcagttgaaa
tctggaactg cctctgttgt gtgcctgctg 480aataacttct atcccagaga ggccaaagta
cagtggaagg tggataacgc cctccaatcg 540ggtaactccc aggagagtgt cacagagcag
gacagcaagg acagcaccta cagcctcagc 600agcaccctga cgctgagcaa agcagactac
gagaaacaca aagtctacgc ctgcgaagtc 660acccatcagg gcctgagctc gcccgtcaca
aagagcttca acaggggaga gtgtggtgcc 720agctccggta tggctgatcc gaaccgtttc
cgcggtaagg acctggcata actcgaggct 780gatcctctac gccggacgca tcgtggccct
agtacgcaag ttcacgtaaa aagggtaact 840agaggttgag gtgattttat gaaaaagaat
atcgcatttc ttcttgcatc tatgttcgtt 900ttttctattg ctacaaacgc gtacgctgag
gttcagctgg tggagtctgg cggtggcctg 960gtgcagccag ggggctcact ccgtttgtcc
tgtgcagctt ctggcttcac cattactaat 1020tccgatatac actgggtgcg tcaggccccg
ggtaagggcc tggaatgggt tgctactatt 1080tatccttatg gcggctatac ttactatgcc
gatagcgtca agggccgttt cactataagc 1140gcagacacat ccaaaaacac agcctaccta
caaatgaaca gcttaagagc tgaggacact 1200gccgtctatt attgtgcaag agggggcggg
atggacggct acgttatgga ctactggggt 1260caaggaacac tagtcaccgt ctcctcggcc
tccaccaagg gcccatcggt cttccccctg 1320gcaccctcct ccaagagcac ctctgggggc
acagcggccc tgggctgcct ggtcaaggac 1380tacttccccg aaccggtgac ggtgtcgtgg
aactcaggcg ccctgaccag cggcgtgcac 1440accttcccgg ctgtcctaca gtcctcagga
ctctactccc tcagcagcgt ggtgaccgtg 1500ccctccagca gcttgggcac ccagacctac
atctgcaacg tgaatcacaa gcccagcaac 1560accaaggtcg acaagaaagt tgagcccaaa
tcttgtgaca aaactcacgg ccgcatgaaa 1620cagctagagg acaaggtcga agagctactc
tccaagaact accacctaga gaatgaagtg 1680gcaagactca aaaaacttgt cggggagcgc
ggaaagctta gtggcggtgg ctctggttcc 1740ggtgattttg attatgaaaa gatggcaaac
gctaataagg gggctatgac cgaaaatgcc 1800gatgaaaacg cgctacagtc tgacgctaaa
ggcaaacttg attctgtcgc tactgattac 1860ggtgctgcta tcgatggttt cattggtgac
gtttccggcc ttgctaatgg taatggtgct 1920actggtgatt ttgctggctc taattcccaa
atggctcaag tcggtgacgg tgataattca 1980cctttaatga ataatttccg tcaatattta
ccttccctcc ctcaatcggt tgaatgtcgc 2040ccttttgtct ttagcgctgg taaaccatat
gaattttcta ttgattgtga caaaataaac 2100ttattccgtg gtgtctttgc gtttctttta
tatgttgcca cctttatgta tgtattttct 2160acgtttgcta acatactgcg taataaggag
tcttaa 2196104256PRTArtificial
SequenceDescription of Artificial Sequence Synthetic polypeptide
104Met Lys Lys Asn Ile Ala Phe Leu Leu Ala Ser Met Phe Val Phe Ser1
5 10 15Ile Ala Thr Asn Ala Tyr
Ala Ser Asp Ile Gln Met Thr Gln Ser Pro 20 25
30Ser Ser Leu Ser Ala Ser Val Gly Asp Arg Val Thr Ile
Thr Cys Arg 35 40 45Ala Ser Gln
Asp Val Ser Thr Ala Val Ala Trp Tyr Gln Gln Lys Pro 50
55 60Gly Lys Ala Pro Lys Leu Leu Ile Tyr Ser Ala Ser
Phe Leu Tyr Ser65 70 75
80Gly Val Pro Ser Arg Phe Ser Gly Ser Gly Ser Gly Thr Asp Phe Thr
85 90 95Leu Thr Ile Ser Ser Leu
Gln Pro Glu Asp Phe Ala Thr Tyr Tyr Cys 100
105 110Gln Gln Ser Tyr Thr Thr Pro Pro Thr Phe Gly Gln
Gly Thr Lys Val 115 120 125Glu Ile
Lys Arg Thr Val Ala Ala Pro Ser Val Phe Ile Phe Pro Pro 130
135 140Ser Asp Glu Gln Leu Lys Ser Gly Thr Ala Ser
Val Val Cys Leu Leu145 150 155
160Asn Asn Phe Tyr Pro Arg Glu Ala Lys Val Gln Trp Lys Val Asp Asn
165 170 175Ala Leu Gln Ser
Gly Asn Ser Gln Glu Ser Val Thr Glu Gln Asp Ser 180
185 190Lys Asp Ser Thr Tyr Ser Leu Ser Ser Thr Leu
Thr Leu Ser Lys Ala 195 200 205Asp
Tyr Glu Lys His Lys Val Tyr Ala Cys Glu Val Thr His Gln Gly 210
215 220Leu Ser Ser Pro Val Thr Lys Ser Phe Asn
Arg Gly Glu Cys Gly Ala225 230 235
240Ser Ser Gly Met Ala Asp Pro Asn Arg Phe Arg Gly Lys Asp Leu
Ala 245 250
255105445PRTArtificial SequenceDescription of Artificial Sequence
Synthetic polypeptide 105Met Lys Lys Asn Ile Ala Phe Leu Leu Ala Ser
Met Phe Val Phe Ser1 5 10
15Ile Ala Thr Asn Ala Tyr Ala Glu Val Gln Leu Val Glu Ser Gly Gly
20 25 30Gly Leu Val Gln Pro Gly Gly
Ser Leu Arg Leu Ser Cys Ala Ala Ser 35 40
45Gly Phe Thr Ile Asn Asn Tyr Asp Ile His Trp Val Arg Gln Ala
Pro 50 55 60Gly Lys Gly Leu Glu Trp
Val Gly Tyr Ile Ser Pro Pro Ser Gly Ala65 70
75 80Thr Tyr Tyr Ala Asp Ser Val Lys Gly Arg Phe
Thr Ile Ser Ala Asp 85 90
95Thr Ser Lys Asn Thr Ala Tyr Leu Gln Met Asn Ser Leu Arg Ala Glu
100 105 110Asp Thr Ala Val Tyr Tyr
Cys Ala Arg Met Val Gly Met Arg Arg Gly 115 120
125Val Met Asp Tyr Trp Gly Gln Gly Thr Leu Val Thr Val Ser
Ser Ala 130 135 140Ser Thr Lys Gly Pro
Ser Val Phe Pro Leu Ala Pro Ser Ser Lys Ser145 150
155 160Thr Ser Gly Gly Thr Ala Ala Leu Gly Cys
Leu Val Lys Asp Tyr Phe 165 170
175Pro Glu Pro Val Thr Val Ser Trp Asn Ser Gly Ala Leu Thr Ser Gly
180 185 190Val His Thr Phe Pro
Ala Val Leu Gln Ser Ser Gly Leu Tyr Ser Leu 195
200 205Ser Ser Val Val Thr Val Pro Ser Ser Ser Leu Gly
Thr Gln Thr Tyr 210 215 220Ile Cys Asn
Val Asn His Lys Pro Ser Asn Thr Lys Val Asp Lys Lys225
230 235 240Val Glu Pro Lys Ser Cys Asp
Lys Thr His Gly Arg Met Lys Gln Leu 245
250 255Glu Asp Lys Val Glu Glu Leu Leu Ser Lys Asn Tyr
His Leu Glu Asn 260 265 270Glu
Val Ala Arg Leu Lys Lys Leu Val Gly Glu Arg Gly Lys Leu Ser 275
280 285Gly Gly Gly Ser Gly Ser Gly Asp Phe
Asp Tyr Glu Lys Met Ala Asn 290 295
300Ala Asn Lys Gly Ala Met Thr Glu Asn Ala Asp Glu Asn Ala Leu Gln305
310 315 320Ser Asp Ala Lys
Gly Lys Leu Asp Ser Val Ala Thr Asp Tyr Gly Ala 325
330 335Ala Ile Asp Gly Phe Ile Gly Asp Val Ser
Gly Leu Ala Asn Gly Asn 340 345
350Gly Ala Thr Gly Asp Phe Ala Gly Ser Asn Ser Gln Met Ala Gln Val
355 360 365Gly Asp Gly Asp Asn Ser Pro
Leu Met Asn Asn Phe Arg Gln Tyr Leu 370 375
380Pro Ser Leu Pro Gln Ser Val Glu Cys Arg Pro Phe Val Phe Ser
Ala385 390 395 400Gly Lys
Pro Tyr Glu Phe Ser Ile Asp Cys Asp Lys Ile Asn Leu Phe
405 410 415Arg Gly Val Phe Ala Phe Leu
Leu Tyr Val Ala Thr Phe Met Tyr Val 420 425
430Phe Ser Thr Phe Ala Asn Ile Leu Arg Asn Lys Glu Ser
435 440 4451062196DNAArtificial
SequenceDescription of Artificial Sequence Synthetic polynucleotide
106atgaaaaaga atatcgcatt tcttcttgca tctatgttcg ttttttctat tgctacaaat
60gcctatgcat ccgatatcca gatgacccag tccccgagct ccctgtccgc ctctgtgggc
120gatagggtca ccatcacctg ccgtgccagt caggatgtgt ccactgctgt agcctggtat
180caacagaaac caggaaaagc tccgaagctt ctgatttact cggcatcctt cctctactct
240ggagtccctt ctcgcttctc tggtagcggt tccgggacgg atttcactct gaccatcagc
300agtctgcagc cggaagactt cgcaacttat tactgtcagc aatcttatac tactcctccc
360acgttcggac agggtaccaa ggtggagatc aaacgaactg tggctgcacc atctgtcttc
420atcttcccgc catctgatga gcagttgaaa tctggaactg cctctgttgt gtgcctgctg
480aataacttct atcccagaga ggccaaagta cagtggaagg tggataacgc cctccaatcg
540ggtaactccc aggagagtgt cacagagcag gacagcaagg acagcaccta cagcctcagc
600agcaccctga cgctgagcaa agcagactac gagaaacaca aagtctacgc ctgcgaagtc
660acccatcagg gcctgagctc gcccgtcaca aagagcttca acaggggaga gtgtggtgcc
720agctccggta tggctgatcc gaaccgtttc cgcggtaagg acctggcata actcgaggct
780gatcctctac gccggacgca tcgtggccct agtacgcaag ttcacgtaaa aagggtaact
840agaggttgag gtgattttat gaaaaagaat atcgcatttc ttcttgcatc tatgttcgtt
900ttttctattg ctacaaacgc gtacgctgag gttcagctgg tggagtctgg cggtggcctg
960gtgcagccag ggggctcact ccgtttgtcc tgtgcagctt ctggcttcac cattaataat
1020tatgatatac actgggtgcg tcaggccccg ggtaagggcc tggaatgggt tggttatatt
1080tctcctccta gcggcgctac ttactatgcc gatagcgtca agggccgttt cactataagc
1140gcagacacat ccaaaaacac agcctaccta caaatgaaca gcttaagagc tgaggacact
1200gccgtctatt attgtgcaag aatggtcggc atgcggaggg gggttatgga ctactggggt
1260caaggaacac tagtcaccgt ctcctcggcc tccaccaagg gcccatcggt cttccccctg
1320gcaccctcct ccaagagcac ctctgggggc acagcggccc tgggctgcct ggtcaaggac
1380tacttccccg aaccggtgac ggtgtcgtgg aactcaggcg ccctgaccag cggcgtgcac
1440accttcccgg ctgtcctaca gtcctcagga ctctactccc tcagcagcgt ggtgaccgtg
1500ccctccagca gcttgggcac ccagacctac atctgcaacg tgaatcacaa gcccagcaac
1560accaaggtcg acaagaaagt tgagcccaaa tcttgtgaca aaactcacgg ccgcatgaaa
1620cagctagagg acaaggtcga agagctactc tccaagaact accacctaga gaatgaagtg
1680gcaagactca aaaaacttgt cggggagcgc ggaaagctta gtggcggtgg ctctggttcc
1740ggtgattttg attatgaaaa gatggcaaac gctaataagg gggctatgac cgaaaatgcc
1800gatgaaaacg cgctacagtc tgacgctaaa ggcaaacttg attctgtcgc tactgattac
1860ggtgctgcta tcgatggttt cattggtgac gtttccggcc ttgctaatgg taatggtgct
1920actggtgatt ttgctggctc taattcccaa atggctcaag tcggtgacgg tgataattca
1980cctttaatga ataatttccg tcaatattta ccttccctcc ctcaatcggt tgaatgtcgc
2040ccttttgtct ttagcgctgg taaaccatat gaattttcta ttgattgtga caaaataaac
2100ttattccgtg gtgtctttgc gtttctttta tatgttgcca cctttatgta tgtattttct
2160acgtttgcta acatactgcg taataaggag tcttaa
2196107255PRTArtificial SequenceDescription of Artificial Sequence
Synthetic polypeptide 107Met Lys Lys Asn Ile Ala Phe Leu Leu Ala Ser
Met Phe Val Phe Ser1 5 10
15Ile Ala Thr Asn Ala Tyr Ala Asp Ile Gln Met Thr Gln Ser Pro Ser
20 25 30Ser Leu Ser Ala Ser Val Gly
Asp Arg Val Thr Ile Thr Cys Arg Ala 35 40
45Ser Gln Asp Val Ser Thr Ala Val Ala Trp Tyr Gln Gln Lys Pro
Gly 50 55 60Lys Ala Pro Lys Leu Leu
Ile Tyr Ser Ala Ser Phe Leu Tyr Ser Gly65 70
75 80Val Pro Ser Arg Phe Ser Gly Ser Gly Ser Gly
Thr Asp Phe Thr Leu 85 90
95Thr Ile Ser Ser Leu Gln Pro Glu Asp Phe Ala Thr Tyr Tyr Cys Gln
100 105 110Gln Ser Tyr Thr Thr Pro
Pro Thr Phe Gly Gln Gly Thr Lys Val Glu 115 120
125Ile Lys Arg Thr Val Ala Ala Pro Ser Val Phe Ile Phe Pro
Pro Ser 130 135 140Asp Glu Gln Leu Lys
Ser Gly Thr Ala Ser Val Val Cys Leu Leu Asn145 150
155 160Asn Phe Tyr Pro Arg Glu Ala Lys Val Gln
Trp Lys Val Asp Asn Ala 165 170
175Leu Gln Ser Gly Asn Ser Gln Glu Ser Val Thr Glu Gln Asp Ser Lys
180 185 190Asp Ser Thr Tyr Ser
Leu Ser Ser Thr Leu Thr Leu Ser Lys Ala Asp 195
200 205Tyr Glu Lys His Lys Val Tyr Ala Cys Glu Val Thr
His Gln Gly Leu 210 215 220Ser Ser Pro
Val Thr Lys Ser Phe Asn Arg Gly Glu Cys Gly Ala Ser225
230 235 240Ser Gly Met Ala Asp Pro Asn
Arg Phe Arg Gly Lys Asp Leu Ala 245 250
255108250PRTArtificial SequenceDescription of Artificial
Sequence Synthetic polypeptide 108Met Lys Lys Asn Ile Ala Phe Leu
Leu Ala Ser Met Phe Val Phe Ser1 5 10
15Ile Ala Thr Asn Ala Tyr Ala Glu Val Gln Leu Val Glu Ser
Gly Gly 20 25 30Gly Leu Val
Gln Pro Gly Gly Ser Leu Arg Leu Ser Cys Ala Ala Ser 35
40 45Gly Phe Thr Ile Ser Gly Ser Trp Ile His Trp
Val Arg Gln Ala Pro 50 55 60Gly Lys
Gly Leu Glu Trp Val Ala Trp Ile Ala Pro Tyr Ser Gly Ala65
70 75 80Thr Asp Tyr Ala Asp Ser Val
Lys Gly Arg Phe Thr Ile Ser Ala Asp 85 90
95Thr Ser Lys Asn Thr Ala Tyr Leu Gln Met Asn Ser Leu
Arg Ala Glu 100 105 110Asp Thr
Ala Val Tyr Tyr Cys Ala Arg Glu Gly Gly Leu Tyr Trp Val 115
120 125Phe Asp Tyr Trp Gly Gln Gly Thr Leu Val
Thr Val Ser Ser Ala Ser 130 135 140Thr
Lys Gly Pro Ser Val Phe Pro Leu Ala Pro Ser Ser Lys Ser Thr145
150 155 160Ser Gly Gly Thr Ala Ala
Leu Gly Cys Leu Val Lys Asp Tyr Phe Pro 165
170 175Glu Pro Val Thr Val Ser Trp Asn Ser Gly Ala Leu
Thr Ser Gly Val 180 185 190His
Thr Phe Pro Ala Val Leu Gln Ser Ser Gly Leu Tyr Ser Leu Ser 195
200 205Ser Val Val Thr Val Pro Ser Ser Ser
Leu Gly Thr Gln Thr Tyr Ile 210 215
220Cys Asn Val Asn His Lys Pro Ser Asn Thr Lys Val Asp Lys Lys Val225
230 235 240Glu Pro Lys Ser
Cys Asp Lys Thr His Leu 245
2501096620DNAArtificial SequenceDescription of Artificial Sequence
Synthetic polynucleotide 109gaattcaact tctccatact ttggataagg
aaatacagac atgaaaaatc tcattgctga 60gttgttattt aagcttgccc aaaaagaaga
agagtcgaat gaactgtgtg cgcaggtaga 120agctttggag attatcgtca ctgcaatgct
tcgcaatatg gcgcaaaatg accaacagcg 180gttgattgat caggtagagg gggcgctgta
cgaggtaaag cccgatgcca gcattcctga 240cgacgatacg gagctgctgc gcgattacgt
aaagaagtta ttgaagcatc ctcgtcagta 300aaaagttaat cttttcaaca gctgtcataa
agttgtcacg gccgagactt atagtcgctt 360tgtttttatt ttttaatgta tttgtaacta
gtacgcaagt tcacgtaaaa agggtatgta 420gaggttgagg tgattttatg aaaaagaata
tcgcatttct tcttgcatct atgttcgttt 480tttctattgc tacaaatgcc tatgcagata
tccagatgac ccagtccccg agctccctgt 540ccgcctctgt gggcgatagg gtcaccatca
cctgccgtgc cagtcaggat gtgtccactg 600ctgtagcctg gtatcaacag aaaccaggaa
aagctccgaa gcttctgatt tactcggcat 660ccttcctcta ctctggagtc ccttctcgct
tctctggtag cggttccggg acggatttca 720ctctgaccat cagcagtctg cagccggaag
acttcgcaac ttattactgt cagcaatctt 780atactactcc tcccacgttc ggacagggta
ccaaggtgga gatcaaacga actgtggctg 840caccatctgt cttcatcttc ccgccatctg
atgagcagtt gaaatctgga actgcctctg 900ttgtgtgcct gctgaataac ttctatccca
gagaggccaa agtacagtgg aaggtggata 960acgccctcca atcgggtaac tcccaggaga
gtgtcacaga gcaggacagc aaggacagca 1020cctacagcct cagcagcacc ctgacgctga
gcaaagcaga ctacgagaaa cacaaagtct 1080acgcctgcga agtcacccat cagggcctga
gctcgcccgt cacaaagagc ttcaacaggg 1140gagagtgtgg tgccagctcc ggtatggctg
atccgaaccg tttccgcggt aaggacctgg 1200cataactcga ggctgatcct ctacgccgga
cgcatcgtgg ccctagtacg caagttcacg 1260taaaaagggt aactagaggt tgaggtgatt
ttatgaaaaa gaatatcgca tttcttcttg 1320catctatgtt cgttttttct attgctacaa
acgcgtacgc tgaggttcag ctggtggagt 1380ctggcggtgg cctggtgcag ccagggggct
cactccgttt gtcctgtgca gcttctggct 1440tcaccattag tggttcttgg atacactggg
tgcgtcaggc cccgggtaag ggcctggaat 1500gggttgcttg gattgctcct tatagcggcg
ctactgacta tgccgatagc gtcaagggcc 1560gtttcactat aagcgcagac acatccaaaa
acacagccta cctacaaatg aacagcttaa 1620gagctgagga cactgccgtc tattattgtg
caagagaggg gggcttgtac tgggtgttcg 1680actactgggg tcaaggaaca ctagtcaccg
tctcctcggc ctccaccaag ggcccatcgg 1740tcttccccct ggcaccctcc tccaagagca
cctctggggg cacagcggcc ctgggctgcc 1800tggtcaagga ctacttcccc gaaccggtga
cggtgtcgtg gaactcaggc gccctgacca 1860gcggcgtgca caccttcccg gctgtcctac
agtcctcagg actctactcc ctcagcagcg 1920tggtgaccgt gccctccagc agcttgggca
cccagaccta catctgcaac gtgaatcaca 1980agcccagcaa caccaaggtc gacaagaaag
ttgagcccaa atcttgtgac aaaactcacc 2040tctagagtgg cggtggctct ggttccggtg
atgctcggtt gccgccgggc gttttttatg 2100ctagcgccgc cctatacctt gtctgcctcc
ccgcgttgcg tcgcggtgca tggagccggg 2160ccacctcgac ctgaatggaa gccggcggca
cctcgctaac ggattcacca ctccaagaat 2220tggagccaat caattcttgc ggagaactgt
gaatgcgcaa accaaccctt ggcagaacat 2280atccatcgcg tccgccatct ccagcagccg
cacgcggcgc atctcgggca gcgttgggtc 2340ctggccacgg gtgcgcatga tcgtgctcct
gtcgttgagg acccggctag gctggcgggg 2400ttgccttact ggttagcaga atgaatcacc
gatacgcgag cgaacgtgaa gcgactgctg 2460ctgcaaaacg tctgcgacct gagcaacaac
atgaatggtc ttcggtttcc gtgtttcgta 2520aagtctggaa acgcggaagt cagcgccctg
caccattatg ttccggatct gcatcgcagg 2580atgctgctgg ctaccctgtg gaacacctac
atctgtatta acgaagcgct ggcattgacc 2640ctgagtgatt tttctctggt cccgccgcat
ccataccgcc agttgtttac cctcacaacg 2700ttccagtaac cgggcatgtt catcatcagt
aacccgtatc gtgagcatcc tctctcgttt 2760catcggtatc attaccccca tgaacagaaa
ttccccctta cacggaggca tcaagtgacc 2820aaacaggaaa aaaccgccct taacatggcc
cgctttatca gaagccagac attaacgctt 2880ctggagaaac tcaacgagct ggacgcggat
gaacaggcag acatctgtga atcgcttcac 2940gaccacgctg atgagcttta ccgcaggatc
cggaaattgt aaacgttaat attttgttaa 3000aattcgcgtt aaatttttgt taaatcagct
cattttttaa ccaataggcc gaaatcggca 3060aaatccctta taaatcaaaa gaatagaccg
agatagggtt gagtgttgtt ccagtttgga 3120acaagagtcc actattaaag aacgtggact
ccaacgtcaa agggcgaaaa accgtctatc 3180agggctatgg cccactacgt gaaccatcac
cctaatcaag ttttttgggg tcgaggtgcc 3240gtaaagcact aaatcggaac cctaaaggga
gcccccgatt tagagcttga cggggaaagc 3300cggcgaacgt ggcgagaaag gaagggaaga
aagcgaaagg agcgggcgct agggcgctgg 3360caagtgtagc ggtcacgctg cgcgtaacca
ccacacccgc cgcgcttaat gcgccgctac 3420agggcgcgtc cggatcctgc ctcgcgcgtt
tcggtgatga cggtgaaaac ctctgacaca 3480tgcagctccc ggagacggtc acagcttgtc
tgtaagcgga tgccgggagc agacaagccc 3540gtcagggcgc gtcagcgggt gttggcgggt
gtcggggcgc agccatgacc cagtcacgta 3600gcgatagcgg agtgtatact ggcttaacta
tgcggcatca gagcagattg tactgagagt 3660gcaccatatg cggtgtgaaa taccgcacag
atgcgtaagg agaaaatacc gcatcaggcg 3720ctcttccgct tcctcgctca ctgactcgct
gcgctcggtc gttcggctgc ggcgagcggt 3780atcagctcac tcaaaggcgg taatacggtt
atccacagaa tcaggggata acgcaggaaa 3840gaacatgtga gcaaaaggcc agcaaaaggc
caggaaccgt aaaaaggccg cgttgctggc 3900gtttttccat aggctccgcc cccctgacga
gcatcacaaa aatcgacgct caagtcagag 3960gtggcgaaac ccgacaggac tataaagata
ccaggcgttt ccccctggaa gctccctcgt 4020gcgctctcct gttccgaccc tgccgcttac
cggatacctg tccgcctttc tcccttcggg 4080aagcgtggcg ctttctcata gctcacgctg
taggtatctc agttcggtgt aggtcgttcg 4140ctccaagctg ggctgtgtgc acgaaccccc
cgttcagccc gaccgctgcg ccttatccgg 4200taactatcgt cttgagtcca acccggtaag
acacgactta tcgccactgg cagcagccac 4260tggtaacagg attagcagag cgaggtatgt
aggcggtgct acagagttct tgaagtggtg 4320gcctaactac ggctacacta gaaggacagt
atttggtatc tgcgctctgc tgaagccagt 4380taccttcgga aaaagagttg gtagctcttg
atccggcaaa caaaccaccg ctggtagcgg 4440tggttttttt gtttgcaagc agcagattac
gcgcagaaaa aaaggatctc aagaagatcc 4500tttgatcttt tctacggggt ctgacgctca
gtggaacgaa aactcacgtt aagggatttt 4560ggtcatgaga ttatcaaaaa ggatcttcac
ctagatcctt ttaaattaaa aatgaagttt 4620taaatcaatc taaagtatat atgagtaaac
ttggtctgac agttaccaat gcttaatcag 4680tgaggcacct atctcagcga tctgtctatt
tcgttcatcc atagttgcct gactccccgt 4740cgtgtagata actacgatac gggagggctt
accatctggc cccagtgctg caatgatacc 4800gcgagaccca cgctcaccgg ctccagattt
atcagcaata aaccagccag ccggaagggc 4860cgagcgcaga agtggtcctg caactttatc
cgcctccatc cagtctatta attgttgccg 4920ggaagctaga gtaagtagtt cgccagttaa
tagtttgcgc aacgttgttg ccattgctgc 4980aggcatcgtg gtgtcacgct cgtcgtttgg
tatggcttca ttcagctccg gttcccaacg 5040atcaaggcga gttacatgat cccccatgtt
gtgcaaaaaa gcggttagct ccttcggtcc 5100tccgatcgtt gtcagaagta agttggccgc
agtgttatca ctcatggtta tggcagcact 5160gcataattct cttactgtca tgccatccgt
aagatgcttt tctgtgactg gtgagtactc 5220aaccaagtca ttctgagaat agtgtatgcg
gcgaccgagt tgctcttgcc cggcgtcaac 5280acgggataat accgcgccac atagcagaac
tttaaaagtg ctcatcattg gaaaacgttc 5340ttcggggcga aaactctcaa ggatcttacc
gctgttgaga tccagttcga tgtaacccac 5400tcgtgcaccc aactgatctt cagcatcttt
tactttcacc agcgtttctg ggtgagcaaa 5460aacaggaagg caaaatgccg caaaaaaggg
aataagggcg acacggaaat gttgaatact 5520catactcttc ctttttcaat attattgaag
catttatcag ggttattgtc tcatgagcgg 5580atacatattt gaatgtattt agaaaaataa
acaaataggg gttccgcgca catttccccg 5640aaaagtgcca cctgacgtct aagaaaccat
tattatcatg acattaacct ataaaaatag 5700gcgtatcacg aggccctttc gtcttcaata
caggtagacc tttcgtagag atgtacagtg 5760aaatccccga aattatacac atgactgaag
gaagggagct cgtcattccc tgccgggtta 5820cgtcacctaa catcactgtt actttaaaaa
agtttccact tgacactttg atccctgatg 5880gaaaacgcat aatctgggac agtagaaagg
gcttcatcat atcaaatgca acgtacaaag 5940aaatagggct tctgacctgt gaagcaacag
tcaatgggca tttgtataag acaaactatc 6000tcacacatcg acaaaccaat acaatacagg
tagacctttc gtagagatgt acagtgaaat 6060ccccgaaatt atacacatga ctgaaggaag
ggagctcgtc attccctgcc gggttacgtc 6120acctaacatc actgttactt taaaaaagtt
tccacttgac actttgatcc ctgatggaaa 6180acgcataatc tgggacagta gaaagggctt
catcatatca aatgcaacgt acaaagaaat 6240agggcttctg acctgtgaag caacagtcaa
tgggcatttg tataagacaa actatctcac 6300acatcgacaa accaatacaa tctacaggta
gacctttcgt agagatgtac agtgaaatcc 6360ccgaaattat acacatgact gaaggaaggg
agctcgtcat tccctgccgg gttacgtcac 6420ctaacatcac tgttacttta aaaaagtttc
cacttgacac tttgatccct gatggaaaac 6480gcataatctg ggacagtaga aagggcttca
tcatatcaaa tgcaacgtac aaagaaatag 6540ggcttctgac ctgtgaagca acagtcaatg
ggcatttgta taagacaaac tatctcacac 6600atcgacaaac caatacaatc
6620110233PRTArtificial
SequenceDescription of Artificial Sequence Synthetic polypeptide
110Met Gly Trp Ser Cys Ile Ile Leu Phe Leu Val Ala Thr Ala Thr Gly1
5 10 15Val His Ser Asp Ile Gln
Met Thr Gln Ser Pro Ser Ser Leu Ser Ala 20 25
30Ser Val Gly Asp Arg Val Thr Ile Thr Cys Arg Ala Ser
Gln Asp Val 35 40 45Ser Thr Ala
Val Ala Trp Tyr Gln Gln Lys Pro Gly Lys Ala Pro Lys 50
55 60Leu Leu Ile Tyr Ser Ala Ser Phe Leu Tyr Ser Gly
Val Pro Ser Arg65 70 75
80Phe Ser Gly Ser Gly Ser Gly Thr Asp Phe Thr Leu Thr Ile Ser Ser
85 90 95Leu Gln Pro Glu Asp Phe
Ala Thr Tyr Tyr Cys Gln Gln Ser Tyr Thr 100
105 110Thr Pro Pro Thr Phe Gly Gln Gly Thr Lys Val Glu
Ile Lys Arg Thr 115 120 125Val Ala
Ala Pro Ser Val Phe Ile Phe Pro Pro Ser Asp Glu Gln Leu 130
135 140Lys Ser Gly Thr Ala Ser Val Val Cys Leu Leu
Asn Asn Phe Tyr Pro145 150 155
160Arg Glu Ala Lys Val Gln Trp Lys Val Asp Asn Ala Leu Gln Ser Gly
165 170 175Asn Ser Gln Glu
Ser Val Thr Glu Gln Asp Ser Lys Asp Ser Thr Tyr 180
185 190Ser Leu Ser Ser Thr Leu Thr Leu Ser Lys Ala
Asp Tyr Glu Lys His 195 200 205Lys
Val Tyr Ala Cys Glu Val Thr His Gln Gly Leu Ser Ser Pro Val 210
215 220Thr Lys Ser Phe Asn Arg Gly Glu Cys225
2301115387DNAArtificial SequenceDescription of Artificial
Sequence Synthetic polynucleotide 111ttcgagctcg cccgacattg
attattgact agttattaat agtaatcaat tacggggtca 60ttagttcata gcccatatat
ggagttccgc gttacataac ttacggtaaa tggcccgcct 120ggctgaccgc ccaacgaccc
ccgcccattg acgtcaataa tgacgtatgt tcccatagta 180acgccaatag ggactttcca
ttgacgtcaa tgggtggagt atttacggta aactgcccac 240ttggcagtac atcaagtgta
tcatatgcca agtacgcccc ctattgacgt caatgacggt 300aaatggcccg cctggcatta
tgcccagtac atgaccttat gggactttcc tacttggcag 360tacatctacg tattagtcat
cgctattacc atggtgatgc ggttttggca gtacatcaat 420gggcgtggat agcggtttga
ctcacgggga tttccaagtc tccaccccat tgacgtcaat 480gggagtttgt tttggcacca
aaatcaacgg gactttccaa aatgtcgtaa caactccgcc 540ccattgacgc aaatgggcgg
taggcgtgta cggtgggagg tctatataag cagagctcgt 600ttagtgaacc gtcagatcgc
ctggagacgc catccacgct gttttgacct ccatagaaga 660caccgggacc gatccagcct
ccgcggccgg gaacggtgca ttggaacgcg gattccccgt 720gccaagagtg acgtaagtac
cgcctataga gtctataggc ccaccccctt ggcttcgtta 780gaacgcggct acaattaata
cataacctta tgtatcatac acatacgatt taggtgacac 840tatagaataa catccacttt
gcctttctct ccacaggtgt ccactcccag gtccaactgc 900acctcggttc tatcgattga
attccaccat gggatggtca tgtatcatcc tttttctagt 960agcaactgca actggagtac
attcagatat ccagatgacc cagtccccga gctccctgtc 1020cgcctctgtg ggcgataggg
tcaccatcac ctgccgtgcc agtcaggatg tgtccactgc 1080tgtagcctgg tatcaacaga
aaccaggaaa agctccgaag cttctgattt actcggcatc 1140cttcctctac tctggagtcc
cttctcgctt ctctggtagc ggttccggga cggatttcac 1200tctgaccatc agcagtctgc
agccggaaga cttcgcaact tattactgtc agcaatctta 1260tactactcct cccacgttcg
gacagggtac caaggtggag atcaaacgaa ctgtggctgc 1320accatctgtc ttcatcttcc
cgccatctga tgagcagttg aaatctggaa ctgcttctgt 1380tgtgtgcctg ctgaataact
tctatcccag agaggccaaa gtacagtgga aggtggataa 1440cgccctccaa tcgggtaact
cccaggagag tgtcacagag caggacagca aggacagcac 1500ctacagcctc agcagcaccc
tgacgctgag caaagcagac tacgagaaac acaaagtcta 1560cgcctgcgaa gtcacccatc
agggcctgag ctcgcccgtc acaaagagct tcaacagggg 1620agagtgttaa gcttggccgc
catggcccaa cttgtttatt gcagcttata atggttacaa 1680ataaagcaat agcatcacaa
atttcacaaa taaagcattt ttttcactgc attctagttg 1740tggtttgtcc aaactcatca
atgtatctta tcatgtctgg atcgggaatt aattcggcgc 1800agcaccatgg cctgaaataa
cctctgaaag aggaacttgg ttaggtatct tctgaggcgg 1860aaagaaccag ctgtggaatg
tgtgtcagtt agggtgtgga aagtccccag gctccccagc 1920aggcagaagt atgcaaagca
tgcatctcaa ttagtcagca accaggtgtg gaaagtcccc 1980aggctcccca gcaggcagaa
gtatgcaaag catgcatctc aattagtcag caaccatagt 2040cccgccccta actccgccca
tcccgcccct aactccgccc agttccgccc attctccgcc 2100ccatggctga ctaatttttt
ttatttatgc agaggccgag gccgcctcgg cctctgagct 2160attccagaag tagtgaggag
gcttttttgg aggcctaggc ttttgcaaaa agctgttaac 2220agcttggcac tggccgtcgt
tttacaacgt cgtgactggg aaaaccctgg cgttacccaa 2280cttaatcgcc ttgcagcaca
tccccccttc gccagctggc gtaatagcga agaggcccgc 2340accgatcgcc cttcccaaca
gttgcgtagc ctgaatggcg aatggcgcct gatgcggtat 2400tttctcctta cgcatctgtg
cggtatttca caccgcatac gtcaaagcaa ccatagtacg 2460cgccctgtag cggcgcatta
agcgcggcgg gtgtggtggt tacgcgcagc gtgaccgcta 2520cacttgccag cgccctagcg
cccgctcctt tcgctttctt cccttccttt ctcgccacgt 2580tcgccggctt tccccgtcaa
gctctaaatc gggggctccc tttagggttc cgatttagtg 2640ctttacggca cctcgacccc
aaaaaacttg atttgggtga tggttcacgt agtgggccat 2700cgccctgata gacggttttt
cgccctttga cgttggagtc cacgttcttt aatagtggac 2760tcttgttcca aactggaaca
acactcaacc ctatctcggg ctattctttt gatttataag 2820ggattttgcc gatttcggcc
tattggttaa aaaatgagct gatttaacaa aaatttaacg 2880cgaattttaa caaaatatta
acgtttacaa ttttatggtg cactctcagt acaatctgct 2940ctgatgccgc atagttaagc
caactccgct atcgctacgt gactgggtca tggctgcgcc 3000ccgacacccg ccaacacccg
ctgacgcgcc ctgacgggct tgtctgctcc cggcatccgc 3060ttacagacaa gctgtgaccg
tctccgggag ctgcatgtgt cagaggtttt caccgtcatc 3120accgaaacgc gcgaggcagt
attcttgaag acgaaagggc ctcgtgatac gcctattttt 3180ataggttaat gtcatgataa
taatggtttc ttagacgtca ggtggcactt ttcggggaaa 3240tgtgcgcgga acccctattt
gtttattttt ctaaatacat tcaaatatgt atccgctcat 3300gagacaataa ccctgataaa
tgcttcaata atattgaaaa aggaagagta tgagtattca 3360acatttccgt gtcgccctta
ttcccttttt tgcggcattt tgccttcctg tttttgctca 3420cccagaaacg ctggtgaaag
taaaagatgc tgaagatcag ttgggtgcac gagtgggtta 3480catcgaactg gatctcaaca
gcggtaagat ccttgagagt tttcgccccg aagaacgttt 3540tccaatgatg agcactttta
aagttctgct atgtggcgcg gtattatccc gtgatgacgc 3600cgggcaagag caactcggtc
gccgcataca ctattctcag aatgacttgg ttgagtactc 3660accagtcaca gaaaagcatc
ttacggatgg catgacagta agagaattat gcagtgctgc 3720cataaccatg agtgataaca
ctgcggccaa cttacttctg acaacgatcg gaggaccgaa 3780ggagctaacc gcttttttgc
acaacatggg ggatcatgta actcgccttg atcgttggga 3840accggagctg aatgaagcca
taccaaacga cgagcgtgac accacgatgc cagcagcaat 3900ggcaacaacg ttgcgcaaac
tattaactgg cgaactactt actctagctt cccggcaaca 3960attaatagac tggatggagg
cggataaagt tgcaggacca cttctgcgct cggcccttcc 4020ggctggctgg tttattgctg
ataaatctgg agccggtgag cgtgggtctc gcggtatcat 4080tgcagcactg gggccagatg
gtaagccctc ccgtatcgta gttatctaca cgacggggag 4140tcaggcaact atggatgaac
gaaatagaca gatcgctgag ataggtgcct cactgattaa 4200gcattggtaa ctgtcagacc
aagtttactc atatatactt tagattgatt taaaacttca 4260tttttaattt aaaaggatct
aggtgaagat cctttttgat aatctcatga ccaaaatccc 4320ttaacgtgag ttttcgttcc
actgagcgtc agaccccgta gaaaagatca aaggatcttc 4380ttgagatcct ttttttctgc
gcgtaatctg ctgcttgcaa acaaaaaaac caccgctacc 4440agcggtggtt tgtttgccgg
atcaagagct accaactctt tttccgaagg taactggctt 4500cagcagagcg cagataccaa
atactgtcct tctagtgtag ccgtagttag gccaccactt 4560caagaactct gtagcaccgc
ctacatacct cgctctgcta atcctgttac cagtggctgc 4620tgccagtggc gataagtcgt
gtcttaccgg gttggactca agacgatagt taccggataa 4680ggcgcagcgg tcgggctgaa
cggggggttc gtgcacacag cccagcttgg agcgaacgac 4740ctacaccgaa ctgagatacc
tacagcgtga gcattgagaa agcgccacgc ttcccgaagg 4800gagaaaggcg gacaggtatc
cggtaagcgg cagggtcgga acaggagagc gcacgaggga 4860gcttccaggg ggaaacgcct
ggtatcttta tagtcctgtc gggtttcgcc acctctgact 4920tgagcgtcga tttttgtgat
gctcgtcagg ggggcggagc ctatggaaaa acgccagcaa 4980cgcggccttt ttacggttcc
tggccttttg ctggcctttt gctcacatgt tctttcctgc 5040gttatcccct gattctgtgg
ataaccgtat taccgccttt gagtgagctg ataccgctcg 5100ccgcagccga acgaccgagc
gcagcgagtc agtgagcgag gaagcggaag agcgcccaat 5160acgcaaaccg cctctccccg
cgcgttggcc gattcattaa tccagctggc acgacaggtt 5220tcccgactgg aaagcgggca
gtgagcgcaa cgcaattaat gtgagttacc tcactcatta 5280ggcaccccag gctttacact
ttatgcttcc ggctcgtatg ttgtgtggaa ttgtgagcgg 5340ataacaattt cacacaggaa
acagctatga ccatgattac gaattaa 5387112468PRTArtificial
SequenceDescription of Artificial Sequence Synthetic polypeptide
112Met Gly Trp Ser Cys Ile Ile Leu Phe Leu Val Ala Thr Ala Thr Gly1
5 10 15Ala Tyr Ala Glu Val Gln
Leu Val Glu Ser Gly Gly Gly Leu Val Gln 20 25
30Pro Gly Gly Ser Leu Arg Leu Ser Cys Ala Ala Ser Gly
Phe Thr Ile 35 40 45Ser Gly Ser
Trp Ile His Trp Val Arg Gln Ala Pro Gly Lys Gly Leu 50
55 60Glu Trp Val Ala Trp Ile Ala Pro Tyr Ser Gly Ala
Thr Asp Tyr Ala65 70 75
80Asp Ser Val Lys Gly Arg Phe Thr Ile Ser Ala Asp Thr Ser Lys Asn
85 90 95Thr Ala Tyr Leu Gln Met
Asn Ser Leu Arg Ala Glu Asp Thr Ala Val 100
105 110Tyr Tyr Cys Ala Arg Glu Gly Gly Leu Tyr Trp Val
Phe Asp Tyr Trp 115 120 125Gly Gln
Gly Thr Leu Val Thr Val Ser Ser Ala Ser Thr Lys Gly Pro 130
135 140Ser Val Phe Pro Leu Ala Pro Ser Ser Lys Ser
Thr Ser Gly Gly Thr145 150 155
160Ala Ala Leu Gly Cys Leu Val Lys Asp Tyr Phe Pro Glu Pro Val Thr
165 170 175Val Ser Trp Asn
Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro 180
185 190Ala Val Leu Gln Ser Ser Gly Leu Tyr Ser Leu
Ser Ser Val Val Thr 195 200 205Val
Pro Ser Ser Ser Leu Gly Thr Gln Thr Tyr Ile Cys Asn Val Asn 210
215 220His Lys Pro Ser Asn Thr Lys Val Asp Lys
Lys Val Glu Pro Lys Ser225 230 235
240Cys Asp Lys Thr His Thr Cys Pro Pro Cys Pro Ala Pro Glu Leu
Leu 245 250 255Gly Gly Pro
Ser Val Phe Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu 260
265 270Met Ile Ser Arg Thr Pro Glu Val Thr Cys
Val Val Val Asp Val Ser 275 280
285His Glu Asp Pro Glu Val Lys Phe Asn Trp Tyr Val Asp Gly Val Glu 290
295 300Val His Asn Ala Lys Thr Lys Pro
Arg Glu Glu Gln Tyr Asn Ser Thr305 310
315 320Tyr Arg Val Val Ser Val Leu Thr Val Leu His Gln
Asp Trp Leu Asn 325 330
335Gly Lys Glu Tyr Lys Cys Lys Val Ser Asn Lys Ala Leu Pro Ala Pro
340 345 350Ile Glu Lys Thr Ile Ser
Lys Ala Lys Gly Gln Pro Arg Glu Pro Gln 355 360
365Val Tyr Thr Leu Pro Pro Ser Arg Glu Glu Met Thr Lys Asn
Gln Val 370 375 380Ser Leu Thr Cys Leu
Val Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val385 390
395 400Glu Trp Glu Ser Asn Gly Gln Pro Glu Asn
Asn Tyr Lys Thr Thr Pro 405 410
415Pro Val Leu Asp Ser Asp Gly Ser Phe Phe Leu Tyr Ser Lys Leu Thr
420 425 430Val Asp Lys Ser Arg
Trp Gln Gln Gly Asn Val Phe Ser Cys Ser Val 435
440 445Met His Glu Ala Leu His Asn His Tyr Thr Gln Lys
Ser Leu Ser Leu 450 455 460Ser Pro Gly
Lys4651136126DNAArtificial SequenceDescription of Artificial Sequence
Synthetic polynucleotide 113attcgagctc gcccgacatt gattattgac
tagttattaa tagtaatcaa ttacggggtc 60attagttcat agcccatata tggagttccg
cgttacataa cttacggtaa atggcccgcc 120tggctgaccg cccaacgacc cccgcccatt
gacgtcaata atgacgtatg ttcccatagt 180aacgccaata gggactttcc attgacgtca
atgggtggag tatttacggt aaactgccca 240cttggcagta catcaagtgt atcatatgcc
aagtacgccc cctattgacg tcaatgacgg 300taaatggccc gcctggcatt atgcccagta
catgacctta tgggactttc ctacttggca 360gtacatctac gtattagtca tcgctattac
catggtgatg cggttttggc agtacatcaa 420tgggcgtgga tagcggtttg actcacgggg
atttccaagt ctccacccca ttgacgtcaa 480tgggagtttg ttttggcacc aaaatcaacg
ggactttcca aaatgtcgta acaactccgc 540cccattgacg caaatgggcg gtaggcgtgt
acggtgggag gtctatataa gcagagctcg 600tttagtgaac cgtcagatcg cctggagacg
ccatccacgc tgttttgacc tccatagaag 660acaccgggac cgatccagcc tccgcggccg
ggaacggtgc attggaacgc ggattccccg 720tgccaagagt gacgtaagta ccgcctatag
agtctatagg cccaccccct tggcttcgtt 780agaacgcggc tacaattaat acataacctt
atgtatcata cacatacgat ttaggtgaca 840ctatagaata acatccactt tgcctttctc
tccacaggtg tccactccca ggtccaactg 900cacctcggtt ctatcgattg aattccacca
tgggatggtc atgtatcatc ctttttctag 960tagcaactgc aactggagcg tacgctgagg
ttcagctggt ggagtctggc ggtggcctgg 1020tgcagccagg gggctcactc cgtttgtcct
gtgcagcttc tggcttcacc attagtggtt 1080cttggataca ctgggtgcgt caggccccgg
gtaagggcct ggaatgggtt gcttggattg 1140ctccttatag cggcgctact gactatgccg
atagcgtcaa gggccgtttc actataagcg 1200cagacacatc caaaaacaca gcctacctac
aaatgaacag cttaagagct gaggacactg 1260ccgtctatta ttgtgcaaga gaggggggct
tgtactgggt gttcgactac tggggtcaag 1320gaaccctggt caccgtctcc tcggcctcca
ccaagggccc atcggtcttc cccctggcac 1380cctcctccaa gagcacctct gggggcacag
cggccctggg ctgcctggtc aaggactact 1440tccccgaacc ggtgacggtg tcgtggaact
caggcgccct gaccagcggc gtgcacacct 1500tcccggctgt cctacagtcc tcaggactct
actccctcag cagcgtggtg actgtgccct 1560ctagcagctt gggcacccag acctacatct
gcaacgtgaa tcacaagccc agcaacacca 1620aggtggacaa gaaagttgag cccaaatctt
gtgacaaaac tcacacatgc ccaccgtgcc 1680cagcacctga actcctgggg ggaccgtcag
tcttcctctt ccccccaaaa cccaaggaca 1740ccctcatgat ctcccggacc cctgaggtca
catgcgtggt ggtggacgtg agccacgaag 1800accctgaggt caagttcaac tggtacgtgg
acggcgtgga ggtgcataat gccaagacaa 1860agccgcggga ggagcagtac aacagcacgt
accgggtggt cagcgtcctc accgtcctgc 1920accaggactg gctgaatggc aaggagtaca
agtgcaaggt ctccaacaaa gccctcccag 1980cccccatcga gaaaaccatc tccaaagcca
aagggcagcc ccgagaacca caggtgtaca 2040ccctgccccc atcccgggaa gagatgacca
agaaccaggt cagcctgacc tgcctggtca 2100aaggcttcta tcccagcgac atcgccgtgg
agtgggagag caatgggcag ccggagaaca 2160actacaagac cacgcctccc gtgctggact
ccgacggctc cttcttcctc tacagcaagc 2220tcaccgtgga caagagcagg tggcagcagg
ggaacgtctt ctcatgctcc gtgatgcatg 2280aggctctgca caaccactac acgcagaaga
gcctctccct gtctccgggt aaatgagtgc 2340gacggcccta gagtcgacct gcagaagctt
ggccgccatg gcccaacttg tttattgcag 2400cttataatgg ttacaaataa agcaatagca
tcacaaattt cacaaataaa gcattttttt 2460cactgcattc tagttgtggt ttgtccaaac
tcatcaatgt atcttatcat gtctggatcg 2520atcgggaatt aattcggcgc agcaccatgg
cctgaaataa cctctgaaag aggaacttgg 2580ttaggtacct tctgaggcgg aaagaaccat
ctgtggaatg tgtgtcagtt agggtgtgga 2640aagtccccag gctccccagc aggcagaagt
atgcaaagca tgcatctcaa ttagtcagca 2700accaggtgtg gaaagtcccc aggctcccca
gcaggcagaa gtatgcaaag catgcatctc 2760aattagtcag caaccatagt cccgccccta
actccgccca tcccgcccct aactccgccc 2820agttccgccc attctccgcc ccatggctga
ctaatttttt ttatttatgc agaggccgag 2880gccgcctcgg cctctgagct attccagaag
tagtgaggag gcttttttgg aggcctaggc 2940ttttgcaaaa agctgttaac agcttggcac
tggccgtcgt tttacaacgt cgtgactggg 3000aaaaccctgg cgttacccaa cttaatcgcc
ttgcagcaca tccccccttc gccagttggc 3060gtaatagcga agaggcccgc accgatcgcc
cttcccaaca gttgcgtagc ctgaatggcg 3120aatggcgcct gatgcggtat tttctcctta
cgcatctgtg cggtatttca caccgcatac 3180gtcaaagcaa ccatagtacg cgccctgtag
cggcgcatta agcgcggcgg gtgtggtggt 3240tacgcgcagc gtgaccgcta cacttgccag
cgccctagcg cccgctcctt tcgctttctt 3300cccttccttt ctcgccacgt tcgccggctt
tccccgtcaa gctctaaatc gggggctccc 3360tttagggttc cgatttagtg ctttacggca
cctcgacccc aaaaaacttg atttgggtga 3420tggttcacgt agtgggccat cgccctgata
gacggttttt cgccctttga cgttggagtc 3480cacgttcttt aatagtggac tcttgttcca
aactggaaca acactcaacc ctatctcggg 3540ctattctttt gatttataag ggattttgcc
gatttcggcc tattggttaa aaaatgagct 3600gatttaacaa aaatttaacg cgaattttaa
caaaatatta acgtttacaa ttttatggtg 3660cactctcagt acaatctgct ctgatgccgc
atagttaagc caactccgct atcgctacgt 3720gactgggtca tggctgcgcc ccgacacccg
ccaacacccg ctgacgcgcc ctgacgggct 3780tgtctgctcc cggcatccgc ttacagacaa
gctgtgaccg tctccgggag ctgcatgtgt 3840cagaggtttt caccgtcatc accgaaacgc
gcgaggcagt attcttgaag acgaaagggc 3900ctcgtgatac gcctattttt ataggttaat
gtcatgataa taatggtttc ttagacgtca 3960ggtggcactt ttcggggaaa tgtgcgcgga
acccctattt gtttattttt ctaaatacat 4020tcaaatatgt atccgctcat gagacaataa
ccctgataaa tgcttcaata atattgaaaa 4080aggaagagta tgagtattca acatttccgt
gtcgccctta ttcccttttt tgcggcattt 4140tgccttcctg tttttgctca cccagaaacg
ctggtgaaag taaaagatgc tgaagatcag 4200ttgggtgcac gagtgggtta catcgaactg
gatctcaaca gcggtaagat ccttgagagt 4260tttcgccccg aagaacgttt tccaatgatg
agcactttta aagttctgct atgtggcgcg 4320gtattatccc gtgatgacgc cgggcaagag
caactcggtc gccgcataca ctattctcag 4380aatgacttgg ttgagtactc accagtcaca
gaaaagcatc ttacggatgg catgacagta 4440agagaattat gcagtgctgc cataaccatg
agtgataaca ctgcggccaa cttacttctg 4500acaacgatcg gaggaccgaa ggagctaacc
gcttttttgc acaacatggg ggatcatgta 4560actcgccttg atcgttggga accggagctg
aatgaagcca taccaaacga cgagcgtgac 4620accacgatgc cagcagcaat ggcaacaacg
ttgcgcaaac tattaactgg cgaactactt 4680actctagctt cccggcaaca attaatagac
tggatggagg cggataaagt tgcaggacca 4740cttctgcgct cggcccttcc ggctggctgg
tttattgctg ataaatctgg agccggtgag 4800cgtgggtctc gcggtatcat tgcagcactg
gggccagatg gtaagccctc ccgtatcgta 4860gttatctaca cgacggggag tcaggcaact
atggatgaac gaaatagaca gatcgctgag 4920ataggtgcct cactgattaa gcattggtaa
ctgtcagacc aagtttactc atatatactt 4980tagattgatt taaaacttca tttttaattt
aaaaggatct aggtgaagat cctttttgat 5040aatctcatga ccaaaatccc ttaacgtgag
ttttcgttcc actgagcgtc agaccccgta 5100gaaaagatca aaggatcttc ttgagatcct
ttttttctgc gcgtaatctg ctgcttgcaa 5160acaaaaaaac caccgctacc agcggtggtt
tgtttgccgg atcaagagct accaactctt 5220tttccgaagg taactggctt cagcagagcg
cagataccaa atactgtcct tctagtgtag 5280ccgtagttag gccaccactt caagaactct
gtagcaccgc ctacatacct cgctctgcta 5340atcctgttac cagtggctgc tgccagtggc
gataagtcgt gtcttaccgg gttggactca 5400agacgatagt taccggataa ggcgcagcgg
tcgggctgaa cggggggttc gtgcacacag 5460cccagcttgg agcgaacgac ctacaccgaa
ctgagatacc tacagcgtga gcattgagaa 5520agcgccacgc ttcccgaagg gagaaaggcg
gacaggtatc cggtaagcgg cagggtcgga 5580acaggagagc gcacgaggga gcttccaggg
ggaaacgcct ggtatcttta tagtcctgtc 5640gggtttcgcc acctctgact tgagcgtcga
tttttgtgat gctcgtcagg ggggcggagc 5700ctatggaaaa acgccagcaa cgcggccttt
ttacggttcc tggccttttg ctggcctttt 5760gctcacatgt tctttcctgc gttatcccct
gattctgtgg ataaccgtat taccgccttt 5820gagtgagctg ataccgctcg ccgcagccga
acgaccgagc gcagcgagtc agtgagcgag 5880gaagcggaag agcgcccaat acgcaaaccg
cctctccccg cgcgttggcc gattcattaa 5940tccaactggc acgacaggtt tcccgactgg
aaagcgggca gtgagcgcaa cgcaattaat 6000gtgagttacc tcactcatta ggcaccccag
gctttacact ttatgcttcc ggctcgtatg 6060ttgtgtggaa ttgtgagcgg ataacaattt
cacacaggaa acagctatga ccatgattac 6120gaatta
6126114245PRTArtificial
SequenceDescription of Artificial Sequence Synthetic polypeptide
114Xaa Xaa Xaa Xaa Xaa Xaa Xaa Xaa Xaa Xaa Xaa Xaa Xaa Xaa Xaa Xaa1
5 10 15Xaa Xaa Xaa Leu Leu Xaa
Xaa Xaa Xaa Xaa Xaa Xaa Pro Xaa Xaa Xaa 20 25
30Xaa Xaa Xaa Xaa Lys Xaa Xaa Xaa Xaa Xaa Xaa Xaa Xaa
Xaa Xaa Xaa 35 40 45Xaa Xaa Xaa
Xaa Tyr Xaa Xaa Xaa Cys Xaa Gln Gly Xaa Xaa Gly Xaa 50
55 60Xaa Gly Arg Asp Gly Xaa Pro Gly Xaa Asn Gly Ile
Pro Gly Thr Pro65 70 75
80Gly Ile Pro Gly Arg Asp Gly Xaa Lys Gly Glu Lys Gly Glu Cys Xaa
85 90 95Xaa Glu Xaa Xaa Glu Glu
Xaa Trp Xaa Pro Asn Xaa Lys Gln Cys Xaa 100
105 110Trp Xaa Xaa Leu Asn Tyr Gly Ile Asp Leu Gly Lys
Ile Ala Xaa Cys 115 120 125Thr Phe
Thr Lys Xaa Arg Ser Xaa Ser Ala Leu Arg Val Leu Phe Xaa 130
135 140Gly Ser Leu Arg Leu Lys Cys Xaa Xaa Ala Cys
Cys Gln Arg Trp Tyr145 150 155
160Phe Thr Phe Xaa Gly Ala Glu Cys Xaa Gly Pro Leu Pro Xaa Glu Xaa
165 170 175Ile Xaa Tyr Leu
Xaa Gln Gly Ser Pro Glu Xaa Asn Ser Thr Ile Asn 180
185 190Ile His Arg Thr Ser Xaa Val Glu Gly Leu Cys
Glu Gly Ile Xaa Ala 195 200 205Gly
Leu Val Asp Xaa Xaa Xaa Trp Val Gly Thr Cys Xaa Asp Tyr Pro 210
215 220Xaa Gly Asp Ala Ser Thr Gly Trp Asn Ser
Val Ser Arg Xaa Ile Ile225 230 235
240Glu Glu Leu Pro Lys 24511511PRTArtificial
SequenceDescription of Artificial Sequence Synthetic peptide 115Xaa
Xaa Ile Xaa Pro Xaa Xaa Gly Xaa Thr Xaa1 5
1011614PRTArtificial SequenceDescription of Artificial Sequence
Synthetic peptide 116Cys Xaa Xaa Xaa Gly Gly Xaa Xaa Xaa Xaa Xaa Xaa
Xaa Xaa1 5 1011715PRTUnknownDescription
of Unknown Yeast sequence 117Gly Trp Asn Ser Val Ser Arg Ile Ile Ile Glu
Glu Leu Pro Lys1 5 10 15
User Contributions:
comments("1"); ?> comment_form("1"); ?>Inventors list |
Agents list |
Assignees list |
List by place |
Classification tree browser |
Top 100 Inventors |
Top 100 Agents |
Top 100 Assignees |
Usenet FAQ Index |
Documents |
Other FAQs |
User Contributions:
Comment about this patent or add new information about this topic:
| People who visited this patent also read: | |
| Patent application number | Title |
|---|---|
| 20210232832 | Method for Lane and Roadway Detection |
| 20210232831 | SIDEWALK INFORMATION GENERATION DEVICE, METHOD AND PROGRAM |
| 20210232830 | Vision-Based Indicator Signal Detection Using Spatiotemporal Filtering |
| 20210232829 | OBJECT RECOGNITION DEVICE AND OBJECT RECOGNITION METHOD |
| 20210232828 | SYSTEM AND METHOD FOR AUTOMATED TABLE GAME ACTIVITY RECOGNITION |
 |  |
 |  |
 |  |
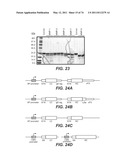 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
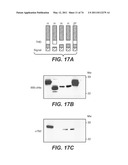 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
| New patent applications in this class: | |
| Date | Title |
|---|---|
| 2022-05-05 | Bispecific antibodies that bind pd-l1 and cd28 |
| 2022-05-05 | Polypeptide modification method for purifying polypeptide multimers |
| 2019-05-16 | Generating targeted sequence diversity in fusion proteins |
| 2019-05-16 | Anti-pd-1 monoclonal antibody and obtaining method therefor |
| 2019-05-16 | Modified chimeric antigen receptors and methods of use |
| New patent applications from these inventors: | |
| Date | Title |
|---|---|
| 2017-02-16 | Treatment of complement-associated disorders |
| 2016-06-30 | Methods of treating fgfr3 related conditions |
| 2015-03-26 | Anti-vegf antibodies |
| 2015-03-05 | Methods of treating liver conditions using notch2 antagonists |
| 2014-10-23 | Compositions and methods for diagnosis and treatment of hepatic cancers |
| Top Inventors for class "Chemistry: natural resins or derivatives; peptides or proteins; lignins or reaction products thereof" | |
| Rank | Inventor's name |
|---|---|
| 1 | Kevin I. Segall |
| 2 | Martin Schweizer |
| 3 | John R. Desjarlais |
| 4 | Brent E. Green |
| 5 | David M. Goldenberg |