Patent application title: Protein secretion in eukaryotic cells
Inventors:
Roland Contreras (Merelbeke, BE)
Steven Geysens (Kruishoutem, BE)
Assignees:
Vlaams Interuniversitar Instituut Voor Biotechnologie VZW
UNIVERSITEIT GENT
IPC8 Class: AC12P2104FI
USPC Class:
435 691
Class name: Chemistry: molecular biology and microbiology micro-organism, tissue cell culture or enzyme using process to synthesize a desired chemical compound or composition recombinant dna technique included in method of making a protein or polypeptide
Publication date: 2009-04-02
Patent application number: 20090087882
Inventors list |
Agents list |
Assignees list |
List by place |
Classification tree browser |
Top 100 Inventors |
Top 100 Agents |
Top 100 Assignees |
Usenet FAQ Index |
Documents |
Other FAQs |
Patent application title: Protein secretion in eukaryotic cells
Inventors:
Roland Contreras
Steven Geysens
Agents:
TRASK BRITT
Assignees:
Origin: SALT LAKE CITY, UT US
IPC8 Class: AC12P2104FI
USPC Class:
435 691
Abstract:
The invention relates to the use of a glucosidase II mutation to increase
protein secretion in yeast cells. The invention relates further to the
use of yeast cells, comprising a mutant glucosidase II gene, possibly in
combination with the expression of a recombinant α-1,2-mannosidase
gene and/or a recombinant N-acetylglucosaminyl-transferase gene, as a
host for protein secretion.Claims:
1. An improvement in a method for secreting exogenous protein from a yeast
cell, the improvement comprising:utilizing a glucosidase II mutation to
increase protein secretion from the yeast cell.
2. The method according to claim 1, wherein said glucosidase II mutation is an inactivating knock out mutation.
3. The method according to claim 1, wherein said glucosidase II mutation is a point mutation.
4. The method according to claim 1, wherein said glucosidase II mutation affects only one subunit of glucosidase II enzyme.
5. The method according to claim 4, wherein said glucosidase II mutation affects subunit alpha.
6. A host for protein secretion, said host comprising:a yeast comprising a recombinant defective glucosidase II.
7. An improvement in a method of the type involving using yeast to secrete protein, the improvement comprising:utilizing, in the method, a yeast having a defective glucosidase II as host for protein secretion.
8. The method according to claim 7 wherein said defective glucosidase II is a recombinant glucosidase II.
9. The method according to claim 7, wherein the yeast is selected from the group consisting of Kluyveromyces sp., Pichia sp., Hansenula sp. or Schizzosaccharomyces pombe.
10. The method according to claim 7, wherein the yeast is a Saccharomyces sp.
11. The method according to claim 7, wherein said glucosidase is defective in subunit alpha.
12. The method according to claim 8, wherein said glucosidase is defective in subunit alpha.
13. The method according to claim 9, wherein said glucosidase is defective in subunit alpha.
14. The method according to claim 10, wherein said glucosidase is defective in subunit alpha.
15. The method according to claim 8, wherein the yeast is selected from the group consisting of Kluyveromyces sp., Pichia sp., Hansenula sp. or Schizzosaccharomyces pombe.
16. The method according to claim 8, wherein the yeast is a Saccharomyces sp.
17. The method according to claim 15, wherein the glucosidase is defective in subunit alpha.
18. The method according to claim 16, wherein the glucosidase is defective in subunit alpha.
19. The method according to claim 2, wherein the glucosidase II mutation affects only one subunit of glucosidase II enzyme.
20. The method according to claim 3 wherein the glucosidase II mutation affects only one subunit of glucosidase II enzyme.
Description:
CROSS-REFERENCE TO RELATED APPLICATIONS
[0001]This application is a continuation of application Ser. No. 11/225,804, filed Sep. 12, 2005, pending, which is a continuation of PCT International Patent Application No. PCT/ep2004/050277, filed on Mar. 10, 2004, designating the United States of America, and published, in English, as PCT International Publication No. WO 2004/081201 A1 on Sep. 23, 2004, which itself claims priority from EP 03075728.0 filed on Mar. 12, 2003, the contents of the entirety of each of which are incorporated by this reference.
TECHNICAL FIELD
[0002]The present invention relates generally to biotechnology, and, more particularly, to the use of a glucosidase II mutation to increase protein secretion in eukaryotic cells. The present invention relates further to the use of eukaryotic cells, comprising a mutant and/or recombinant glucosidase II gene, possibly in combination with the expression of a recombinant α-1,2-mannosidase gene and/or a recombinant N-acetylglucosaminyl-transferase gene, as a host for protein secretion.
BACKGROUND
[0003]Filamentous fungi can produce high yields of proteins and metabolites. Impressive increases in the secretion of homologous proteins were obtained with traditional strain-improvement strategies based on various mutagenesis approaches. As such, industrial strains have been created that secrete>20 g/l of a specific endogenous protein. In this way, filamentous fungi seem promising organisms for the production of heterologous proteins of biomedical interest (Maras et al., 1999; Punt et al., 2002).
[0004]However, unlike mammalian cells, these lower eukaryotic organisms do not synthesize complex type protein-linked oligosaccharides. This inability hampers the use of therapeutic glycoproteins produced by filamentous fungi, since they mostly synthesize high-mannose type N-glycans. Due to the presence of several lectins on human cells, glycoproteins carrying this type of glycosylation are rapidly cleared from the blood stream. This significantly reduces their therapeutic value.
[0005]Not only are lower eukaryotes like filamentous fungi, unable to synthesize complex type oligosaccharides, they sometimes also elongate the high-mannose type glycans with fungal-specific glycan residues like mannosephosphate, α-1,3-mannose and galactofuranose. Some of these residues induce an immunogenic response in humans, again reducing the therapeutic value of such glycoproteins.
[0006]Protein N-glycosylation originates in the endoplasmic reticulum (ER), where an N-linked oligosaccharide (Glc3Man9GlcNAc2) assembled on dolichol (a lipid carrier intermediate) is transferred to the appropriate Asn of a nascent protein. This is a co-translational event common to all eukaryotic organisms. The three glucose residues and one specific α-1,2-linked mannose residue are removed by specific glucosidases and an α-1,2-mannosidase in the ER, resulting in the core oligosaccharide structure, Man8GlcNAc2. Proteins with this core sugar structure are transported to the Golgi apparatus where the sugar moiety undergoes various modifications. Significant differences exist in the modifications of the sugar chain in the Golgi apparatus between lower and higher eukaryotes.
[0007]In mammalian cells, the modification of the sugar chain can follow 3 different pathways depending on the protein moiety to which it is added. That is: (1) the core sugar chain does not change; (2) the core sugar chain is changed by adding the N-acetylglucosamine-1-phosphate moiety (GlcNAc-1-P) in UDP-N-acetyl glucosamine (UDP-GlcNAc) to the 6-position of mannose in the core sugar chain, followed by removal of the GlcNAc moiety to form an acidic sugar chain in the glycoprotein; and (3) the core sugar chain is first converted into Man5GlcNAc2 by removing 3 mannose residues with Golgi a Mannosidase I; Man5GlcNAc2 is then further modified by adding GlcNAc and removing 2 more mannose residues, followed by sequentially adding GlcNAc, galactose (Gal), and N-acetylneuraminic acid (also called sialic acid (NeuNAc)) to form various hybrid or complex sugar chains (R. Kornfeld and S. Kornfeld, 1985; Chiba et al., 1998).
[0008]In filamentous fungi like Trichoderma reesei, only a part of the Man8(9)GlcNAc2 structures are (partially) trimmed down to Man5GlcNAc2. These oligosaccharides can then be further modified to fungal-specific glycans through the addition of mannosephosphate residues in a diester linkage. As such, a variety of sugar residues can be found on Trichoderma secreted glycoproteins, consisting of .sub.Man5-8(9) GlcNAc2 with or without one or two mannosephosphate residues. An exception to this general Trichoderma glycosylation pattern is the Rut-C30 strain, producing mainly GlcMan7(-9)GlcNAc2 or GlcMan7(-9)GlcNAc2-P-Man (Maras et al., 1997).
[0009]A clear need exists for a fungal strain, such as a Trichoderma strain, that is able to secrete large amounts of a heterologous protein with a more human-compatible glycosylation profile. As such, the Rut-C30 strain of T. Reesei which is a hypersecretor of endogenous cellulases (up to 30 g/l), would be an interesting strain for heterologous protein production, but it is hampered by its aberrant glycosylation pattern, compared to the wild type Qm6a strain and to most of the industrial mutant strains. In these Trichoderma strains, a first α-1,2-linked glucose residue is removed by glucosidase I, after transfer of the Glc3Man9GlcNAc2 structure to the protein. This is followed by the removal of the two α-1,3-linked glucose residues by glucosidase II. However in the Rut-C30 strain, NMR analysis revealed that more than 80% of the glycan structures synthesized on cellobiohydrolase I (CBH I) still contained one α-1,3-linked glucose residue at the end of the α-1,3-arm of the high-mannose core structure (Maras et al, 1997). This indicates a malfunction at the level of the glucosidase II. This malfunction could be due to a reduced expression level of the enzyme.
SUMMARY OF THE INVENTION
[0010]Surprisingly, we found that this malfunction is due to the presence of a frameshift mutation within the Rut-C30 glucosidase II ORF, presumably deleting or severely damaging the Glc-α-1,3-Man substrate binding site, but not the Glc-α-1,3-Glc substrate binding site. This presumption would be in accordance with the kinetic model proposed by Alonso et al. (1993), in which the two substrate binding sites are proposed, and could also explain why the removal of the first α-1,3-linked glucose residue does not seem to present any problem.
[0011]Even more surprisingly we found that a Rut-C30 strain expressing a fully functional (ER-localized) T. Reesei glucosidase II was showing a changed glycosylation profile, resembling that of most other T. Reesei strains. However, the secretion level was affected by the expression of the glucosidase II. Coexpression of glucosidase II, α-1,2-mannosidase and GlcNac-transferase resulted in a modified secretion, combined with a human-like glycosylation profile. The resulting strain may be useful for the production of heterologous proteins of which the glycosylation pattern is critical.
[0012]Knocking out the glucosidase II gene in Saccharomyces cerevisiae, as well as the introduction of the mutant glucosidase II form similar to the T. Reesei RUT C30 mutation confirms the unexpected effect of the glucosidase II mutation on the protein secretion.
[0013]Therefore, one embodiment of the invention involves the use of a glucosidase II mutation to increase protein secretion in eukaryotic cells. Every mutation that affects the activity of the glucosidase II may be used, and it may be, as a non-limiting example, an inactivating or downregulating mutation in the promoter region, an inactivating knock out of a part of the coding sequence or of the whole coding sequence, a point mutation in one or more of the subunits of the glucosidase II, or an exchange of one or more of the subunits by a mutant subunit or by a subunit of another species. Preferably, the effect of the mutation is a decrease in activity of glucosidase II. Preferably, the subunit that carries the mutation is subunit alpha.
[0014]The eukaryotic cells may be any eukaryotic cells, including, but not limited to mammalian cells, insect cells, plant cells and fungal cells. Preferably, said eukaryotic cell is a fungal cell, even more preferably a filamentous fungus or a yeast cell. Filamentous fungi are known to the person, skilled in the art, and include, but are not limited to, species from the genera Aspergillus, Fusarium, Geotrichum, Monascus, Monilia, Mucor, Penecillium, Rhizopus, Trichoderma and Ustilago. Preferably, the filamentous fungus is a Trichoderma sp., even more preferably the filamentous fungus is T. Reesei Rut-C30. Yeast cells are also known to the person skilled in the art and include, but are not limited to Saccharomyces sp., Pichia sp., Hansenula sp., Kluyveromyces sp. and Schizosaccharomyces pombe. Preferably, the yeast cell is a Saccharomyces cerevisiae strain.
[0015]The secreted proteins may be homologous proteins or heterologous proteins, and they may be glycosylated or not glycosylated. Preferably, the secreted proteins are heterologous proteins, and even more preferably, the proteins are glycosylated heterologous proteins.
[0016]Another aspect of the invention is the use of a recombinant filamentous fungus comprising a defective recombinant glucosidase II as a host for protein secretion.
[0017]A defective recombinant glucosidase II as used here means that the endogenous sequence of the promoter and/or of the coding sequence of one or more of the subunits of glucosidase II has been replaced by a non-endogenous sequence. Preferably, the subunit that is replaced is subunit alpha. The non-endogenous sequence may be the sequence of a non-glucosidase II gene of the same organism, or the sequence of another organism, or an artificial sequence. The resulting defective recombinant glucosidase II should have an activity that is different from the wild type, preferentially a lower activity. Filamentous fungi are known to the person, skilled in the art, and include, but are not limited to, species from the genera Aspergillus, Fusarium, Geotrichum, Monascus, Monilia, Mucor, Penecillium, Rhizopus, Trichoderma and Ustilago. Preferably, the filamentous fungus is a Trichoderma sp., even more preferably the filamentous fungus is T. Reesei Rut-C30.
[0018]Protein secretion as used here may be the secretion of an endogenous protein, or the secretion of a heterologous protein.
[0019]Still another aspect of the invention is the use of a yeast comprising a defective glucosidase II as a host for protein secretion. The defective glucosidase II has an activity that is different from the wild type, preferably a lower activity. The defective glucosidase II might be obtained by random mutagenesis. However, preferably the defective glucosidase II is a defective recombinant glucosidase II, as discussed above. Yeast cells are preferably selected from the group consisting of Saccharomyces sp., Pichia sp., Hansenula sp., Kluyveromyces sp. and Schizosaccharomyces pombe. Even more preferably, the yeast cell is a Saccharomyces cerevisiae strain.
[0020]Still another aspect of the invention is a method to increase protein secretion of a eukaryotic cell, comprising mutagenesis of glucosidase II. Techniques for mutagenesis are known to the person skilled in the art, and include, but are not limited to chemical mutagenesis, physical mutagenesis such as UV radiation, or site directed mutagenesis by recombinant DNA techniques. Preferably, the mutagenesis is site directed mutagenesis. Preferably, the eukaryotic cell is a fungal cell, such as a filamentous fungus or a yeast cell. Glucosidase II genes have been cloned from a number of mammalian species including rat (Trombetta et al., 1996), mouse (Arendt et al., 1997), pig (Flura et al., 1997) and human (Trombetta et al., 1996, genbank accession number D42041). The glucosidase II protein from these mammalian species consists of an alpha and a beta subunit. The alpha subunit is about 110 kDa and contains the catalytic activity of the enzyme, while the beta subunit has a C-terminal HDEL ER-retention sequence and is believed to be required for the ER localization of the enzyme. Similar results were obtained for the fission yeast S. pombe (d'Alessio et al., 1999). The sequence of the glucosidase II gene from S. cerevisiae has also been identified (ORF YBR229c, located on chromosome II, genbank accession number Z36098). This gene encodes a protein of about 110 kDa, which shows a high degree of homology to the mammalian alpha subunits. During the course of our work, the genes coding for the α-subunits of the T. Reesei Rut-C30 and the Aspergillus niger glucosidase II protein, were cloned, facilitating the site directed mutagenesis of the genes.
[0021]Transformation vectors and transformation techniques for yeast and filamentous fungi are known to the person skilled in the art. For Trichoderma, preferred vectors carrying a glucosidase II expression sequence are called pFGPDglsIITreesei and pFGPDglsIITreesei(Myc).
[0022]Vectors can be introduced into the cells of a Trichoderma strain using known methods such as the protoplast technique, described by Penttila et al., 1987. Other published methods useful for transformation of the plasmids or linear vectors include electroporation (Goldman et al., 1990), particle bombardment (Lorito et al., 1993) and an Agrobacterium tumefaciens-mediated strategy (de Groot et al., 1998).
[0023]During the transformation procedure, the glucosidase II expression sequence is cotransformed with a selection plasmid. By "selection plasmid" is meant a plasmid carrying a selection marker. By "selection marker" is meant an expression cassette coding for a specific gene product, which enables us to discriminate between a transformed strain and a non-transformed strain. Transformed Trichoderma clones can be selected by using appropriate techniques including but not limited to culturing auxotrophic cells after transformation in the absence of the biochemical product required (due to the cell's auxotrophy), selection for and detection of a new phenotype, or culturing in the presence of an antibiotic which is toxic to the fungus in the absence of a resistance gene within the transformants. Examples of available selection markers for T. Reesei are the acetamidase expression cassette of the vector p3SR2 (Hynes et al., 1983) (enabling transformed strains to grow on acetamide as a sole nitrogen source), the E. coli hygromycin B phosphotransferase cassette of vector pAN7.1 (Punt et al., 1987) and the Streptoalloteichus hindustanus phleomycin-binding protein expression cassette of vector pAN8.1 (Mattern et al., 1988) (enabling the transformed strains to grow on a certain concentration of hygromycin resp. phleomycin).
[0024]Another aspect of the invention is a genetically engineered filamentous fungus expressing a glucosidase II gene according to the invention, further expressing a recombinant α-1,2-mannosidase gene. Preferably, the α-1,2-mannosidase gene is fused to an ER retention signal. More preferably, the ER retention signal is derived from the MNS1 protein of S. cerevisiae. Even more preferably, the retention signal comprises the sequence HDEL. Preferably, the filamentous fungus is a Trichoderma sp., even more preferably the filamentous fungus is T. Reesei Rut-C30.
[0025]An α-1,2-mannosidase cleaves the α-1,2-linked mannose residues at the end of Man8(9)GlcNAc2, and converts this core oligosaccharide on glycoproteins to Man5GlcNAc2 which is thought to be a very poor substrate for a Golgi phosphomannosyltransferase. Thus, by introducing an α-1,2-mannosidase into filamentous fungi such as Trichoderma, glycoproteins with reduced mannose and phosphate content can be produced. Furthermore, Man5GlcNAc2 is the acceptor substrate for the mammalian N-acetylglucosaminyl transferase I and as such a key structure in the synthesis of hybrid- and complex-type sugar chains, characteristic for mammalian glycoproteins.
[0026]According to the present invention, a genetically engineered Trichoderma strain capable of expressing an α-1,2-mannosidase can be generated by introducing into the filamentous fungus a nucleotide sequence capable of expressing the α-1,2-mannosidase.
[0027]According to the present invention, the nucleotide sequence encoding an α-1,2-mannosidase for introduction into a Trichoderma strain can derive from any species. A number of α-1,2-mannosidase genes have been cloned from different species and are available to those skilled in the art, including mammalian genes encoding, e.g., a murine α-1,2-mannosidase (Herscovics et al., 1994), a rabbit α-1,2-mannosidase (Lal et al., 1994) or a human α-1,2-mannosidase (Tremblay et al., 1998), as well as fungal genes encoding, e.g., an Aspergillus α-1,2-mannosidase (Eades et al., 1998), a T. Reesei α-1,2-mannosidase (Maras et al., 2000), or a Saccharomyces cerevisiae α-1,2-mannosidase (Camirand et al., 1991). Protein sequence analysis has revealed a high degree of conservation among the eukaryotic α-1,2-mannosidases identified so far.
[0028]Preferably, the nucleotide sequence for introduction into a Trichoderma strain encodes a fungal α-1,2-mannosidase, more preferably, a T. Reesei α-1,2-mannosidase, and more particularly, the T. Reesei α-1,2-mannosidase described by Maras et al., since it is known to also have a broad substrate specificity (Maras et al., 2000; Van Petegem et al., 2001).
[0029]According to the present invention, the nucleotide sequence can encode a full length α-1,2-mannosidase or a functional part thereof. By "functional part" is meant a polypeptide fragment of an α-1,2-mannosidase which substantially retains the enzymatic activity of the full-length protein. By "substantially" is meant that at least about 40%, or preferably, at least 50% or more of the full-length α-1,2-mannosidase activity is retained. Those skilled in the art can readily identify and make functional parts of an α-1,2-mannosidase using a combination of techniques known in the art. Predictions of the portions of an α-1,2-mannosidase essential to or sufficient to confer the enzymatic activity can be made based on analysis of the protein sequence. The activity of a portion of an α-1,2-mannosidase of interest, expressed and purified from an appropriate expression system, can be verified using in vitro or in vivo assays.
[0030]In accordance with the present invention, an α-1,2-mannosidase or a functional part thereof expressed in a Trichoderma strain preferably localizes at a place in the secretory pathway where Man8/9GlcNAc2 (the substrate of α-1,2-mannosidase) is already formed on a glycoprotein, but has not reached the location of the secretion pathway in which resides the phosphomannosyltransferase.
[0031]Accordingly, the α-1,2-mannosidase or a functional part thereof is engineered to include an ER-retention signal, such that the protein expressed in a Trichoderma strain is targeted to the ER and retains therein for function. "An ER retention signal" refers to a peptide sequence, which directs a protein having such peptide sequence to be transported to and retained in the ER. Such ER retention sequences are often found in proteins that reside and function in the ER. Multiple choices of ER retention signals are available to those skilled in the art, e.g., the first 21 amino acid residues of the S. cerevisiae ER protein MNS1 (Martinet et al., 1998). A preferred ER retention signal for use in the present invention is peptide HDEL. The HDEL sequence found at the C-terminus of a number of yeast proteins acts as a retention/retrieval signal for the ER (Pelham, 1988). Proteins with an HDEL sequence are bound by a membrane-bound receptor (Erd2p) and then enter a retrograde transport pathway to return from the Golgi apparatus into the ER. According to the present invention, an ER retention signal can be placed anywhere in the protein sequence of an α-1,2-mannosidase, but preferably at the C-terminal end of the α-1,2-mannosidase.
[0032]The α-1,2-mannosidase for use in the present invention can be further modified, e.g., by insertion of an epitope tag to which antibodies are available, such as Myc, HA, FLAG and His6 tags which are well-known in the art. An epitope-tagged α-1,2-mannosidase can be conveniently monitored for both expression and intracellular localization. An ER retention signal and an epitope tag can be readily introduced into a protein of interest by inserting nucleotide sequences coding for such signal or tag into the nucleotide sequence encoding the protein of interest, using any of the molecular biology techniques known in the art.
[0033]According to the present invention, the nucleotide sequence coding for an α-1,2-mannosidase or a functional part thereof can be placed in an operable linkage to a promoter and a 3' termination sequence.
[0034]Promoters appropriate for expression of an α-1,2-mannosidase in a Trichoderma strain can include both constitutive promoters and inducible promoters. Constitutive promoters include e.g., the Aspergillus nidulans glyceraldehyde-3-phosphate dehydrogenase promoter ("the gpdA promoter"). Examples of inducible promoters include, e.g., the T. Reesei cellobiohydrolase I promoter ("the CBHI promoter").
[0035]Transcription termination sequences are sequences 3' to the stop codon of a structural gene which function to stabilize the mRNA transcription product of the gene to which the sequence is operably linked, such as sequences which elicit polyadenylation. Examples of such 3' termination sequences are the T. Reesei cellobiohydrolase I terminator ("the CBHI terminator") and the A. nidulans indoleglycerolphosphate synthase terminator ("TrypC terminator").
[0036]The preferred vector carrying an α-1,2-mannosidase expression sequence is called pFGPDGLAT3-MFManHDEL.
[0037]Vectors can be introduced into the cells of a Trichoderma strain using known methods such as the protoplast technique, described by Penttila et al., 1987. Other published methods useful for transformation of the plasmids or linear vectors include electroporation (Goldman et al., 1990), particle bombardment (Lorito et al, 1993) and an Agrobacterium tumefaciens-mediated strategy (de Groot et al., 1998).
[0038]During the transformation procedure, the α-1,2-mannosidase expression sequence is cotransformed with a selection plasmid. By "selection plasmid" is meant a plasmid carrying a selection marker. By "selection marker" is meant an expression cassette coding for a specific gene product, which enables us to discriminate between a transformed strain and a non-transformed strain. Transformed Trichoderma clones can be selected by using appropriate techniques including but not limited to culturing auxotrophic cells after transformation in the absence of the biochemical product required (due to the cell's auxotrophy), selection for and detection of a new phenotype, or culturing in the presence of an antibiotic which is toxic to the fungus in the absence of a resistance gene within the transformants. Examples of available selection markers for T. Reesei are the acetamidase expression cassette of the vector p3SR2 (Hynes et al., 1983) (enabling transformed strains to grow on acetamide as a sole nitrogen source), the E. coli hygromycin B phosphotransferase cassette of vector pAN7.1 (Punt et al., 1987) and the Streptoalloteichus hindustanus phleomycin-binding protein expression cassette of vector pAN8.1 (Mattern et al., 1988) (enabling the transformed strains to grow on a certain concentration of hygromycin resp. phleomycin).
[0039]A further aspect of the invention is a genetically engineered filamentous fungus, expressing a glucosidase II gene according to the invention, further expressing a recombinant N-acetylglucosaminyl-transferase I gene (GlcNAc-transferase I or GnTI). Preferably, the GnTI gene is a human gene. Even more preferably, the GnTI gene is fused to a Golgi localization signal, preferably a Golgi localization signal derived from a protein with SEQ ID NO:4, even more preferably a Golgi localization signal comprising SEQ ID NO:5, even more preferably a Golgi localization signal essentially consisting of SEQ ID NO:5, most preferably a Golgi localization signal consisting of SEQ ID NO:5. Preferably, the filamentous fungus is a Trichoderma sp., even more preferably the filamentous fungus is T. Reesei Rut-C30.
[0040]A GlcNAc-Transferase I is responsible for the addition of β-1,2-GlcNAc to Man5GlcNAc2, and converts this core oligosaccharide on glycoproteins to GlcNAcMan5GlcNAc2. The mannose residues of GlcNAcMan5GlcNAc2 can be further trimmed by a mammalian Golgi mannosidase II. The resulting GlcNAcMan3GlcNAc2 structure can be further elongated with other glycan residues to form hybrid or complex type sugar branches characteristic of mammalian glycoproteins. Thus, by way of introducing a GlcNAc-transferase I into filamentous fungi such as T. Reesei, glycoproteins with a mammalian-like or cognate glycoprotein pattern can be produced.
[0041]According to the present invention, the nucleotide sequence encoding a GlcNAc-transferase I (GnTI) for use in the expression vector of the present invention can derive from any higher eukaryotic species, e.g., rabbit (Sarkar et al., 1991; SWISS-PROT Accession No P27115), human (Schachter, 1991; SWISS-PROT Accession No P26572), rat (Fukuda et al., 1994; SWISS-PROT Accession No Q09325), plants and insects. Preferably, the nucleotide sequence for use in the present vectors encodes a human GnTI. More preferably, the GnTI gene comprises SEQ ID NO:1, even more preferably, the GnTI gene is essentially consisting of SEQ ID NO:1, most preferably, the GnTI gene is consisting of SEQ ID NO:1.
[0042]According to the present invention, the nucleotide sequence can also encode only a functional part of a GlcNAc-Transferase I. By "functional part" is meant a polypeptide fragment of a GlcNAc-Transferase I, which substantially retains the enzymatic activity of the full-length protein. By "substantially" is meant at least about 40%, or preferably, at least 50% or more of the enzymatic activity of the full-length GlcNAc-Transferase I is retained. For example, as illustrated by the present invention, the catalytic domain of the human GnTI constitutes a "functional part" of the human GnTI. Those skilled in the art can readily identify and make functional parts of a GlcNAc-Transferase I using a combination of techniques known in the art. Predictions of the portions of a GlcNAc-Transferase I essential to, or sufficient to confer the enzymatic activity can be made based on analysis of the protein sequence. The activity of a portion of a GlcNAc-Transferase I of interest, expressed and purified from an appropriate expression system, can be verified using in vitro or in vivo assays.
[0043]In accordance with the present invention, a GnTI or a functional part thereof expressed in a T. Reesei strain preferably is targeted to a site in the secretory pathway where Man5GlcNAc2 (the substrate of GnTI) is already formed on a glycoprotein. Preferably, the GnTI or a functional part is targeted to the Golgi apparatus.
[0044]Accordingly, in a preferred embodiment of the present invention, the GnTI is engineered as such that the GnTI or a functional part thereof expressed from the vector is fused with a fungal Golgi localization signal. "A fungal Golgi localization signal" refers to a peptide sequence, which directs a protein having such a peptide sequence to be retained in the Golgi apparatus. Such Golgi localization sequences are often found in proteins that reside and function in the Golgi apparatus. Choices of Golgi localization signals are available to those skilled in the art. A preferred Golgi localization signal for use in the present invention is a peptide derived from the N-terminal part of a Saccharomyces cerevisiae Kre2 protein (ScKre2). According to the present invention, a Golgi localization signal can be placed anywhere within the GnTI, but preferably at the terminus of the GnTI, and more preferably at the N-terminus of the GnTI.
[0045]The GnTI for use in the present invention can be further modified, e.g., by insertion of an epitope tag to which antibodies are available, such as Myc, HA, FLAG and His6 tags well known in the art. An epitope-tagged GnTI can be conveniently purified, or monitored for both expression and intracellular localization. A Golgi localization signal and an epitope tag can be readily introduced into a protein of interest by inserting nucleotide sequences coding for such signal or tag into the nucleotide sequence encoding the protein of interest, using any of the molecular biology techniques known in the art.
[0046]According to the present invention, the nucleotide sequence coding for a GlcNAc transferase I or a functional part thereof can be placed in an operable linkage to a promoter and a 3' termination sequence.
[0047]Promoters appropriate for expression of a GlcNAc transferase I in a Trichoderma strain can include both constitutive promoters and inducible promoters. Constitutive promoters include e.g., the Aspergillus niger glyceraldehyde-3-phosphate dehydrogenase promoter ("the gpdA promoter"). Examples of inducible promoters include, e.g., the T. Reesei cellobiohydrolase I promoter ("the CBHI promoter").
[0048]3' termination sequences are sequences 3' to the stop codon of a structural gene which function to stabilize the mRNA transcription product of the gene to which the sequence is operably linked, such as sequences which elicit polyadenylation. Examples of such 3' termination sequences are the T. Reesei cellobiohydrolase I terminator ("the CBHI terminator") and the A. nidulans indoleglycerolphosphate synthase terminator ("TrypC terminator").
[0049]The preferred vector carrying a GlcNAc transferase I expression sequence is called pFGPDKrecohGnTI.
[0050]Vectors can be introduced into the cells of a Trichoderma strain using known methods such as the protoplast technique, described by Penttila et al., 1987. Other published methods useful for transformation of the plasmids or linear vectors include electroporation (Goldman et al., 1990), particle bombardment (Lorito et al, 1993) and an Agrobacterium tumefaciens-mediated strategy (de Groot et al., 1998).
[0051]During the transformation procedure, the GlcNAc transferase I expression sequence is cotransformed with a selection plasmid. By "selection plasmid" is meant a plasmid carrying a selection marker. By "selection marker" is meant an expression cassette coding for a specific gene product, which enables us to discriminate between a transformed strain and a non-transformed strain. Transformed Trichoderma clones can be selected by using appropriate techniques including but not limited to culturing auxotrophic cells after transformation in the absence of the biochemical product required (due to the cell's auxotrophy), selection for and detection of a new phenotype, or culturing in the presence of an antibiotic which is toxic to the fungus in the absence of a resistance gene within the transformants. Examples of available selection markers for T. Reesei are the acetamidase expression cassette of the vector p3SR2 (Hynes et al., 1983) (enabling transformed strains to grow on acetamide as a sole nitrogen source), the E. coli hygromycin B phosphotransferase cassette of vector pAN7.1 (Punt et al., 1987) and the Streptoalloteichus hindustanus phleomycin-binding protein expression cassette of vector pAN8.1 (Mattern et al., 1988) (enabling the transformed strains to grow on a certain concentration of hygromycin resp. phleomycin).
[0052]Another aspect of the invention is a filamentous fungus expressing a recombinant glucosidase II gene, according to the invention, further expressing both a recombinant α-1,2-mannosidase gene and a recombinant GlcNAc-transferase I gene.
[0053]Still another aspect of the invention is the use of a genetically modified filamentous fungus, according to the invention, to modulate protein secretion, compared with the parental strain.
BRIEF DESCRIPTION OF THE SEVERAL VIEWS OF THE DRAWINGS
[0054]The patent or application file contains at least one drawing executed in color. Copies of this patent or patent application publication with color drawing(s) will be provided by the Office upon request and payment of the necessary fee.
[0055]FIG. 1: Schematic overview of the inverse PCR strategy.
[0056]FIG. 2: Construction strategy for the glucosidase II expression plasmids pFGPDglsIITreesei and pFGPDglsIITreeseiMyc.
[0057]FIG. 3: (A) PCR on Rut-C30 cDNA library using degenerate primers 1 and 3. (B) nested PCR on Rut-C30 cDNA library using degenerate primers 1, 2 and 3.
[0058]FIG. 4: PCR screening with degenerate primers 1 and 3: (A) first round with about 5000 clones per well; (B) second round with about 500 clones per well; (C) third round with about 50 clones per well. The cell suspension from well A2 was used for the second PCR round; the cell suspension from well F3 was used for the third PCR round and the cell suspension from well B9 was used for colony hybridization analysis.
[0059]FIG. 5: results of the colony hybridization.
[0060]FIG. 6: plasmid DNA of the 7 positive clones was prepared and digested with XhoI/EcoRI to isolate the cDNA insert. Hybridization analysis indicates that at least the 1700 bp fragment is glucosidase II specific.
[0061]FIG. 7: (A) cloning of the 5' part of the glsII ORF by inverse PCR; (B) cloning of the 5' part of the glsII ORF by RACE; (C) sequence comparison between the inverse PCR and the 5' RACE fragment reveals the existence of an intron region.
[0062]FIG. 8: Glycosylation profile of the RutC30, QM9414 and g14 transformants, either native, after α-1,2-mannosidase digestion or after mild acid hydrolysis. For all three cases it is clear that the g14 transformant has a glycosylation profile that contains characteristics of both the RutC30 and the QM9414 strains. The deduced N-glycans are numbered: 1: Man5GlcNAc2; 2: Man6GlcNAc2; 3: Man8GlcNAc2; 4: Man9GlcNAc2; 5: GlcMan7GlcNAc2; 6: GlcMan9GlcNAc2; 7: GlcMan9GlcNAc2; 3': ManPMan8GlcNAc2; 4': ManPMan9GlcNAc2; 5': ManPGlcMan7GlcNAc2; 5'': PGlcMan7GlcNAc2.
[0063]FIG. 9: Southern blot analysis of the genomic DNA of several hygromycin resistant transformants and of the WT RutC30 strain: transformant g14 expresses both the mutant and the repaired glucosidase II alpha-subunit gene; all other transformants grow on hygromycin but have not integrated the repaired glucosidase II alpha-subunit gene into the genome. The fragment of 3400 bp (arrow) indicates the random integration of the glucosidase II expression cassette. The fragment of 5000 bp represents the hybridization signal against the endogenous mutant glucosidase II alpha subunit gene. Ref: PstI digested lambda DNA as reference.
[0064]FIG. 10: Construction strategy for the α-1,2-mannosidase expression plasmid pFGPDLAT3-MFManHDEL.
[0065]FIG. 11: Construction strategy for the GlcNAc transferase I expression plasmid pFGPDKrecoGnTI.
[0066]FIG. 12: N-glycan analysis of several transformants capable of growing on acetamide as single N-source: transformants F4, F17, F18 and F32 almost exclusively synthesize Man5GlcNAc2 as a result of the expression of an ER-localized α-1,2-mannosidase.
[0067]FIG. 13: PCR analysis of the genomic DNA of a few acetamide resistant Trichoderma clones: Transformants F4, F17, F18 en F32 score positive for the PCR analysis.
[0068]FIG. 14: Probability of coiled coil structure as predicted by the paircoil algorithm. A: predicted coiled coil of GnTI, the maximal probability is 0.36. B: predicted coiled coil of yeast Kre2, maximal probability 0.69.
[0069]FIG. 15: Each gel represents separate experiments in which the secretion level of the g14 transformant and the RutC30 wild type strain were compared with one another. For each analysis, the different protein samples were prepared from different but simultaneously grown cultures of both strains. In the first gel, Hygr1 and Hygr2 represent hygromycin resistant RutC30 transformants that have no genomic integration of the full-size glucosidase II (checked on gDNA and via N-glycan analysis). As a result, they have a similar secretion behavior as the untransformed RutC30 strain.
[0070]FIG. 16: Strategy for the construction of a S. cerevisiae rot2 knock out, and for the consequent replacement of the URA3 cassette by a mutant glucosidase II gene, carrying the RutC30 T. reesei glucosidase II mutation
[0071]FIG. 17: DSA-FACE analysis of the rot2 knock out transformants (KO16, KO18, KO20) as confirmed by PCR, in comparison with a transformant with an aberrant PCR pattern (KO11) and the parental strain YA-72, and with the rot2 knock out mutant Y13369 and its parental strain BY4742. All rot2 knock outs show a similar sugar pattern that is clearly different from that of the wild type strains.
[0072]FIG. 18: IFNβ-specific Western blot of proteins secreted in the medium by 8 BY4742 IFNβ producing clones (1-8) and 8 Y13369 IFNβ producing clones (A-H). M: marker; WT: non-transformed BY4742 parental strain; KO: non-transformed Y13369 rot2 knock out mutant. The average OD600 value of the cultures was 12.56 for the BY4742 transformants and 12.65 for the Y13369 transformants. The upper band is the glycosylated form, the lower band is the not glycosylated form.
[0073]FIG. 19: IFNβ specific Western blot of pooled medium proteins from cultures of 8 BY4742 IFNβ producing clones (WT), 8 Y13369 IFNβ producing clones (KO) and 8 Y13369 IFNβ producing clones supertransformed with pYX132LEUGLSIImut3' (mut). M: marker, C1 and C2: untransformed parental strains. The upper band is the glycosylated form, the lower band is the not glycosylated form.
DETAILED DESCRIPTION OF THE INVENTION
[0074]The invention is further explained with the aid of the following illustrative Examples.
EXAMPLES
Materials and methods
Strains and Transformation Procedure:
[0075]Two T. Reesei strains were used for the glucosidase II work, being the Rut-C30 (ATCC 56765) and the QM9414 (ATCC 26921) strain. Trichoderma transformations were by co-transformation according to Penttila et al. (1987) using the hygromycin resistance gene (plasmid pAN7.1 (Punt et al., 1987)) as a selection marker. Before transformation, the glucosidase II expression vectors pFGPDglsIITreesei and pFGPDglsIITreeseiMyc were linearized with FspI (Biolabs). Transformants were selected on minimal medium (composition per liter: 20 g dextrose monohydrate, 5 g (NH4)2SO4, 15 g KH2PO4, 0.3 g CaCl2, 0.3 g MgSO4 and mineral components) containing 150 μg/ml of hygromycin.
[0076]T. Reesei QM9414 was used for the expression of an ER-localized α-1,2-mannosidase. Transformation was by co-transformation according to Penttila et al. (1987) using AmdS (plasmid p3SR2, Hynes et al., 1983)) as a selection marker. Before transformation, the α-1,2-mannosidase expression plasmid was linearized with NdeI (Biolabs). Transformants were selected on minimal medium with acetamidase as the sole nitrogen source (composition per liter: 20 g dextrose monohydrate, 15 g KH2PO4, 0.3 g CaCl2, 0.3 g MgSO4, mineral components, 10 ml 1M acetamidase and 12.5 ml 1M CsCl).
[0077]T. Reesei QM9414-F4 was used for the expression of a Golgi-localized GlcNAc-transferase I. This strain is a functional α-1,2-mannosidase transformant of strain QM9414. Transformation was by co-transformation according to Penttila et al. (1987) using the phleomycin resistance gene as a selection marker. Before transformation, the GlcNAc-transferase I expression plasmid was linearized with NdeI (Biolabs). Transformants were selected on minimal medium (composition per liter: 20 g dextrose monohydrate, 15 g KH2PO4, 5 g (NH4)2SO4, 0.3 g CaCl2, 0.3 g MgSO4, mineral components) containing 150 μg/ml zeocin (Invitrogen).
[0078]For the cloning of the glucosidase II gene and for the construction work, we used electrocompetent resp. chemocompetent E. coli MC1061 cells (hsdR2 hsdM.sup.+ hsdS.sup.+ araD139 .sub.Δ(ara leu)7697ΔlacX74 galE15 galK16 rpsL (Strr) mcrA mcrB1) (Casadaban et al., 1980). Growth and transformations were as described in Sambrook et al., (1989).
[0079]Saccharomyces cerevisiae YA-72 (MATa, his3, ura3, leu2) is an IFN-β producing yeast strain, obtained by transforming the strain CL3-ABYS86 with a GAL1-MF(IS)-IFNβ-CYCT integrative expression cassette (Demolder et al., 1994).
[0080]S. cerevisiae Y13369 is a rot2 knock out (MATα,his3, leu2, ura3, YBR229c::kanMX4) from BY4742 and was obtained from EUROFAN. The parental strain BY4742 was used as reference.
[0081]Yeast strains were transformed using the LiAc method.
Nucleic Acid Preparations from Filamentous Fungi
[0082]Trichoderma genomic DNA was prepared from 5 to 6 day old mycelium, grown in shake flasks in minimal medium (composition per liter: 20 g dextrose monohydrate, 5 g (NH4)2SO4, 15 g KH2PO4, 0.3 g CaCl2, 0.3 g MgSO4 and mineral components) at 30° C. The mycelium was separated from the growth medium and grinded using liquid nitrogen. 5 to 10 ml of extraction buffer (200 mM Tris.HCl pH 8.5; 250 mM NaCl; 0.5% SDS) was added to resuspend the disrupted Trichoderma cells. An equal amount of a phenol/chloroform/isoamyl alcohol mixture (25/24/1) was added to the suspension. After mixing, samples were centrifuged for 1 hour at 2500 g. The upper phase, containing the DNA, was transferred to a new tube and incubated with 1 mg of RNaseA for 30 minutes at 37° C. Following a new extraction with an equal volume of a mixture of chloroform/isoamyl alcohol (24/1), the upper phase was transferred to a new tube. The DNA was precipitated with half a volume of isopropanol (centrifugation at full speed for 20 min. at 4° C.). After removing the supernatant, the DNA pellet was washed with 70% EtOH, dried at 37° C. and resuspended in a suitable volume of H2O.
[0083]Total Trichoderma RNA was prepared from 5 to 6 day old mycelium, grown in shake flasks in minimal medium at 30° C. The mycelium was separated from the growth medium and grinded using liquid nitrogen. Per 0.2 g of mycelium, 1 ml of extraction buffer (25 mM sodiumcitrate; 4 M GuHCl; 100 mM sodium-lauryl sarcosine and 100 mM beta-mercapto-ethanol) was added. The suspension was thoroughly mixed and incubated at 50° C. for 15 minutes. An equal amount of a phenol/chloroform/isoamyl alcohol mixture (25/24/1) was added to the suspension. After mixing, samples were centrifuged for 15 minutes at 9000 g and 4° C. This extraction was repeated twice and followed by a chloroform/isoamyl alcohol (24/1) extraction. After centrifugation (4° C., 9000 g, 15 minutes), the upper phase was collected. One volume of 6 M LiCl, was added and the RNA was precipitated overnight at 4° C. After centrifugation (4° C., 9000 g, 15 minutes), the obtained RNA pellet was resuspended in one volume of 3 M LiCl and again precipitated through centrifugation (4° C., 9000 g, 15 minutes). The pellet was resuspended in 400 μl of 0.3 M NaOAc pH 5.7 and incubated at 50° C. for 10 minutes. After centrifugation (4° C., 9000 g, 15 minutes), the supernatant was collected. 1 ml of ice-cold EtOH was added and the RNA was precipitated overnight at -20° C. The suspension was centrifuged at 4° C. for 20 minutes and the obtained pellet was washed with 70% EtOH. The dried pellet was resuspended in a suitable volume of DEPC treated H2O.
Cloning of the Trichoderma Glucosidase II Gene
[0084]Cloning of the glucosidase II alpha subunit was initiated from a T. Reesei Rut-C30 cDNA library (Merja Penttila, VTT Biotechnology). In this library, which contains about 100,000 clones, the Rut-C30 cDNA was cloned into an EcoRI/XhoI opened pAJ401 yeast expression vector (Saloheimo et al., 1994).
[0085]Based on the alignment of several known mammalian and yeast alpha subunit amino acid sequences, three homologous regions were selected on which degenerate primers were synthesized: the sense primer 5'-GTITATGGIATHCCIGAGCATGC-3' (SEQ ID NO:6) and the antisense primers 5'-GIGCGTGIGCICKGAAGAAIG-3' (SEQ ID NO:7), and 5'-TGISWICCIGCGAAGAAIGCIC-3' (SEQ ID NO:8), with H=A, C and T; K=G and T; S=G and C; W=A and T. Amplification with primers 1/3 and with primers 1/2 should result in a DNA fragment of approximately 1170 resp. 970 bp. Reaction conditions for the amplification with primers 1/3 were the following: 94° C. for 45 sec.; 55° C. for 1 min., 72° C. for 1.5 min. Similar reaction conditions were used for the nested PCR, except for the annealing temperature which was decreased to 50° C. Obtained PCR fragments were cloned into pCR2.1-TOPO (Invitrogen) for sequence analysis. TOPO-cloning was done as described by the manufacturer.
[0086]As a screening strategy for a bacterial clone containing the T. reesei glucosidase II alpha subunit, we used the technique of "Rapid cDNA cloning by PCR screening" (Takumi and Lodish, 1994). In brief, the cDNA library was transformed to E. coli MC1061 competent cells. The transformation mixture was diluted and divided into a 96 well plate in a way that every well contained about 5000 cDNA clones. As such, the whole microtiter plate represented about 5 times the number of cDNA clones within the library. After incubation for several hours at 37° C., a PCR was performed with primers 1 and 3 as described above on cellular mixtures of the 12 columns and the 8 rows of the 96 well plate. Based on these results positive wells, lying on the crossing of positive columns and positive rows, could be identified. The cell suspension of one of the positive wells was inoculated into the wells of a new microtiter plate at 500 clones per well. The PCR strategy was repeated and the cell suspension of one of the resulting positive wells was again inoculated into the wells of a new microtiter plate, this time at a concentration of 50 clones per well. By using the PCR strategy, again new positive wells were identified. From one of these wells, the cell suspension was plated on solid Luria Bertani medium. About 200 colonies were transferred to Hybond N filters (Amersham), incubated overnight and analyzed through colony hybridization using the Trichoderma glucosidase II specific 1170 bp PCR fragment as probe. 32P-labeling of the probe was done using the High-Prime kit (Roche), following the instructions of the manufacturer.
[0087]DNA was prepared from several positive clones and digested with EcoRI (Gibco BRL) and XhoI (Gibco BRL) to release the cDNA insert. The glucosidase II specificity of the obtained fragments was checked by southern blotting, using the 32P-labeled 1170 bp PCR fragment as probe. Also, the obtained fragments were cloned for sequence analysis either as an EcoRI/XhoI fragment into an EcoRI/SalI (Roche) opened pUC19 vector or as a blunted XhoI fragment into an EcoRV (Gibco BRL) opened pBluescriptII KS +/- (Stratagene) vector.
5'-RACE and Inverse PCR
[0088]To clone the 5' missing part of the glucosidase II alpha subunit gene, both 5'-RACE and inverse PCR were used. For the inverse PCR (iPCR) strategy, an antisense (5'-GTTAAACGTTTCGTCCCACC-3') (SEQ ID NO:9) and sense (5'-GGCTCCATCCCTTTCATGC-3') (SEQ ID NO:10) PCR primer were designed, based on the 5' sequence of the cloned but incomplete glucosidase II alpha subunit Rut-C30 cDNA. The 5' end of the primers is facing each other and hybridizes to positions on the cDNA that are separated by 229 bp containing an NcoI restriction site. 10 μg of genomic Trichoderma DNA was digested at 37° C. for several hours with 100 units BamHI (Gibco BRL), a restriction enzyme that cuts the cloned cDNA sequence, 3' to both iPCR primers. After heat inactivation of BamHI (10 minutes at 65° C.), the obtained genomic DNA fragments were induced to self-circulate through overnight incubation at room temperature in the presence of 5 units T4 DNA ligase (Roche). Following a phenol extraction and isopropanol precipitation, the DNA was digested with 50 units NcoI (Biolabs) for several hours at 37 C.°. As such, the desired glucosidase II containing genomic DNA fragment will be linearized again, enabling the designed iPCR primers (now facing each other with their 3' ends) to hybridize each to one end of the fragment. Following a new phenol extraction and isopropanol precipitation, the DNA was resuspended into 50 μl of H2O. 1 μl of this DNA suspension was used as a template in a PCR reaction with 50 pmol of each iPCR primer. The PCR reaction was performed with cloned Pfu polymerase (Stratagene) in a total volume of 100 μl, and consisted of 20 cycli of 94° C. for 45 sec.; 55° C. for 30 sec. and 72° C. for 1 min. 30 sec. A schematic overview of the inversed PCR strategy is shown in FIG. 1.
[0089]For the 5'-RACE procedure, we made use of the First Choice® RLM-RACE strategy kit from Ambion. Primer design and experimental procedure was done on total RNA, following the instructions of the manufacturer. For the outer PCR primer ROT2TR-RLMRACE (5'-GATATACTCGAAGACGTCGG-3') (SEQ ID NO:11) was used. For the inner PCR, we used primer ROT2TR4_AS (5'-GTTAAACGTTTCGTCCCACC-3') (SEQ ID NO:9). Annealing during the outer PCR reaction was performed at 57° C.; for the inner PCR, a temperature of 55° C. was used.
[0090]The 5'-RACE and inverse PCR fragments were cloned into the pCR-blunt II-TOPO vector (Invitrogen) for sequence analysis, following the instructions of the manufacturer.
Intron and Frame-Shift Analysis through PCR
[0091]The intron-exon composition of the glucosidase II gene was analyzed by amplifying the whole gene from the Rut-C30 genome. 1 μg of gDNA was used as template; the sequence of the sense resp. antisense primer was 5'-ATGAGGTCGACGATGGGG-3' (SEQ ID NO:12) resp. 5'-AGCCAGCTTGATGCTCC-3' (SEQ ID NO:13). Using Pfu polymerase (Stratagene), following reaction conditions were applied: 25 cycles of 94° C. for 1 min.; 55° C. for 1 min. and 72° C. for 7 min.
[0092]Frame-shift analysis was done by PCR on the Rut-C30 and QM9414 genome. 1 μg of gDNA was used as PCR template. The sequence of the internal glucosidase II specific primers was 5'-TATCTCTGGTTTCCCGTTCTCG-3' (SEQ ID NO:14) for the sense primer ROT2TR3--5 and 5'-CTGGTCATCAATCGCCAAGCC-3' (SEQ ID NO:15) for the antisense primer ROT2TR0_AS. PCR was performed using Pfu polymerase and following reaction conditions: 25 cycles of 94° C. for 1 min.; 60° C. for 1 min. and 72° C. for 1 min.
[0093]The PCR fragments were cloned into the pCR-blunt II-TOPO vector (Invitrogen) for sequence analysis, following the instructions of the manufacturer.
Construction of the Trichoderma Expression Vector for a Functional Trichoderma glucosidase II Alpha Subunit Gene
[0094]In a first step, the cloned glucosidase II cDNA fragment was cut out of the pAJ401 library vector as an approximately 3000 bp EcoRI/HindIII (GibcoBRL) fragment. This fragment was ligated into an EcoRI/HindIII opened pUC19 vector, resulting in plasmid pUC19.sub.ΔglsIITreesei(shift). In a second step, the frame-shift within the cloned Rut-C30 cDNA fragment was repaired. Using genomic DNA from the QM9414 strain as a template and Pfu polymerase (Stratagene), a PCR reaction was started with primers ROT2TR2_S (5'-ATCAATGAGCAACTCCTGGC-3') (SEQ ID NO:16) and ROT2TR0_AS (5'-CTGGTCATCAATCGCCAAGCC-3') (SEQ ID NO:15). The PCR reaction went on for 25 cycli of 1 min. at 95° C., 1 min. at 60° C. and 1 min. at 72° C. The obtained fragment was digested with XcmI (Biolabs)/PflMI (Biolabs) and ligated into the XcmI/PflMI opened vector pUC19.sub.ΔglsIITreesei(shift), resulting into the vector pUC19.sub.ΔglsIITreesei (repaired). In a third step, the ORF of the glucosidase II alpha subunit was completed: for this the 5'RACE fragment (materials and methods 4) was digested with DraIII (Biolabs) and MspAI (Biolabs) and ligated into the DraIII/EcoRI-Klenow (Roche) treated vector pUC19.sub.ΔglsIITreesei (repaired), resulting into the plasmid pUC19glsIITreesei. In a next step, a unique SmaI site was incorporated at the 3' terminus of the glucosidase II ORF through mutagenesis, using the Quick Change Mutagenesis kit form Stratagene. The primer couple used to induce the silent mutation (from CGT to CGG) consisted of a sense primer 5'-CCATGTGAAGGCCCGGGTTGGGGATGACTGG-3' (SEQ ID NO:17) and an antisense primer 5'-CCAGTCATCCCCAACCCGGGCCTTCACATGG-3' (SEQ ID NO:18). The resulting plasmid was called pUC19glsIITreesei(SmaI). In a following step, the plasmid was cut EcoRI/SalI for the integration of a linker at the 5' end of the glucosidase II ORF. The linker consisted of two partially complementary primers (sense primer: 5'-GAATTCCCGCGGTACGTAATTATGAGG-3' (SEQ ID NO:19) and antisense primer: 5'GTCGACCTCATAATTACGTACCGCGGG-3') (SEQ ID NO:20) and was prepared by mixing both primers, boiling the mixture and gradually cooling it to room temperature. By inserting the linker, two new and unique restriction sites (SacII and SnaBI) were integrated at the 5' end of the glucosidase II ORF, creating plasmid pUC19(5')glsIITreesei(SmaI). In a next step, this plasmid was opened HindIII/SacII-T4 (Roche) treated and ligated into the HindIII/NcoI(Biolabs)-S1(Gibco BRL) treated plasmid pFGPDGLAT3 (Contreras et al., 1991). As such the glucosidase II alpha subunit ORF was placed under the transcriptional control of the constitutive A. nidulans gpdA promoter. To decrease the distance between the 3' end of the ORF and the TrpC terminator, the vector was digested with MluI (Gibco BRL) to remove a fragment of about 500 bp. The obtained vector fragment was closed by overnight ligation, resulting in the plasmid pFGPDglsIITreesei. A variant of this plasmid was constructed, containing the Trichoderma glucosidase II ORF with a C-terminal Myc-tag. For this, vector pUC19(5')glsIITreesei(SmaI) was digested with SmaI (Gibco BRL) and SnaBI (Biolabs). The resulting fragment containing most of the glucosidase II ORF, was ligated into an NcoI (S1 treated)/Bsp120I (MBI Fermentas) (Klenow treated) opened pFGPDglsIIScMyc vector. Using this construction strategy the 10 C-terminal amino acids of the Trichoderma glucosidase II were replaced by the coding sequence for the Myc-tag. In the resulting vector, called pFGPDglsIITreeseiMyc, the ORF coding for the Myc-tagged Trichoderma glucosidase II alpha subunit is under the transcriptional control of the constitutive A. nidulans gpdA promoter and the TrpC terminator. Plasmid pFGPDglsIIScMyc was constructed for the expression of the S. cerevisiae glucosidase II alpha subunit in T. Reesei. This vector was constructed as follows: by a PCR strategy using plasmid pGAPZglsIIScMyc as DNA template, Pfu polymerase, sense primer ROT2ScNco_S 5'-CTTGCCATGGTCCTTTTGAAATGGCTC-3' (SEQ ID NO:21) and antisense primer ROT2ScMycHind_AS 5'-CCCAAGCTTCTACAGATCCTCTTCTGAGATGAG-3' (SEQ ID NO:22), we amplified a Myc-tagged version of the S. cerevisiae glucosidase II gene. The PCR reaction consisted of 30 cycli of 45 sec. at 94° C., 45 sec. at 50° C. and 8 min. at 72° C. Since the nucleotide sequences of the NcoI and HindIII restriction sites were incorporated in the sense resp. antisense primer, the obtained PCR fragment was easily cloned into an NcoI/HindIII opened pFGPDGLAT2 vector, resulting into plasmid pFGPDglsIIScMyc. Vector pGAPZglsIIScMyc was constructed for the expression of the S. cerevisiae glucosidase II ORF in Pichia pastoris (PCT International Patent Application WO0200856). Genomic DNA was prepared form the S. cerevisiae strain InvSC1(α, leu2-3, 112 his3Δ1, trp1-289, ura3-52) (Invitrogen) using the Nucleon kit (Amersham). This was used as template for the amplification of the glucosidase II alpha subunit with sense primer ROT2Sc_S 5'-CCGCTCGAGATGGTCCTTTTGAAATGGCTC-3' (SEQ ID NO:23) (containing the sequence for a unique XhoI restriction site) and antisense primer ROT2Sc_AS 5'-CCGGGCCCAAAAATAACTTCCCAATCTTCA-3' (SEQ ID NO:24) (containing the sequence for a unique ApaI restriction site). Amplification was performed by a touch-down PCR strategy using LA TaKaRa polymerase (TaKaRa Shuzo co., LTD.) with following conditions: 3 cycli of 30 sec. at 94° C., 2 sec. at 98° C., 30 sec. at 65° C. and 10 min. at 70° C.; 3 cycli of 30 sec. at 94° C., 2 sec. at 98° C., 30 sec. at 60° C. and 10 min. at 70° C. and 30 cycli of 30 sec. at 94° C., 2 sec. at 98° C., 30 sec. at 55° C. and 10 min. at 70° C. After digestion with ApaI (Biolabs)/XhoI (Gibco BRL), the fragment was ligated into an ApaI/XhoI opened pGAPZ,A vector (Invitrogen), to allow in frame cloning of the amplified glucosidase II ORF with a nucleotide sequence coding for the Myc-tag. The resulting plasmid was called pGAPZglsIIScMyc. An overview of the construction strategy can be seen in FIG. 2.
Construction of the α-1,2-Mannosidase and GlcNAc-Transferase Expression Plasmids
[0095]For the expression of an ER-localized variant of the T. Reesei α-1,2-mannosidase in T. Reesei Rut-C30-g31, the α-1,2-mannosidase coding part was isolated from plasmid pGAPZMFManHDEL. This plasmid contains the mannosidase with the N-terminal prepro-signal sequence of the S. cerevisiae α-mating factor and a C-terminal HDEL-tag as described in Callewaert et al. (2001b). The mannosidase part was isolated by a BstBI (Biolabs)/NotI (Biolabs) digest. The BstBI sticky end was blunted with T4-polymerase (Roche). The obtained fragment was ligated in an NcoI (Biolabs) (Mung Bean nuclease (Roche) blunted)/NotI opened pFGPDGLAT3 (Contreras et al., 1991) vector. The resulting plasmid pFGPDGLAT3-MFManHDEL contains the α-1,2-mannosidase ORF under the transcriptional control of a constitutive gpdA promoter. An overview of the construction scheme is presented in FIG. 11.
[0096]In order to target more efficiently the human GlcNAc-transferase I to the fungal Golgi apparatus, the GnTI N-terminal part was replaced by the S. cerevisiae Kre2 N-terminal sequence, known to be responsible for protein retention in the yeast Golgi (Lussier, et al., 1995). Plasmid pFGPDKrecoGnTI was constructed as follows. Plasmid YEp352Kre2 (kindly provided by Dr. Howard Bussey, McGill University, Montreal, Canada), which contains the Kre2 gene as a SacI/PvuII fragment cloned in a SalI(Klenow blunted)/SacI opened YEp352 vector, was digested with SacI (Biolabs)/PvuI (Gibco BRL) and T4-polymerase (Roche) blunted. The 5' end region of the gene was isolated and cloned in a Klenow blunted SgrAI (Roche)/XbaI (Gibco BRL) opened pUChGnTI vector (Maras et al., 1997). By doing so, the coding sequence of the Golgi localization signal of the yeast Kre2 protein was cloned in frame with the nucleotide sequence of the catalytic domain of the GlcNAc transferase I protein. The resulting ORF was isolated by performing an EcoRV (Gibco BRL)/HindIII (Gibco BRL) double digest and was cloned into an NcoI (S1-nuclease (Gibco BRL) blunted)/HindIII opened pFGPDGLAT3 vector, as such creating the plasmid pFGPDKrecoGnTI. The construction of the expression plasmid is presented in FIG. 11.
Genomic Analysis
[0097]For the analysis of the glucosidase II transformants, genomic DNA was digested overnight with 50 units of NheI (Biolabs) and KpnI. After electrophoresis, the DNA was transferred to a Hybond N.sup.+ membrane (Amersham). Integration of the expression plasmid into the genome was checked, by hybridizing the Hybond filter with a 32P-labeled glucosidase II-specific probe. Labeling of the probe was done using the High Prime kit (Roche). The DNA template for the labeling reaction consisted of a part of the glucosidase II ORF and was obtained through an NcoI digest on plasmid pFGPDglsIITreesei.
[0098]A similar strategy was followed after digestion of the genomic DNA with 50 u of DraIII and BglII (Biolabs). This time however, the Southern blot was screened with a probe which is derived from an EcoRI/NheI fragment of vector pFGPDglsIITreesei and which hybridizes against the gpdA promoter sequence of the glucosidase II expression plasmid.
[0099]For the analysis of the α-1,2-mannosidase transformants, genomic DNA was digested overnight with 50 units BglII (Promega) and NotI (Promega). After electrophoresis, the DNA was transferred to a Hybond N.sup.+ membrane (Amersham). Integration of the expression plasmid into the genome was checked by hybridizing the Hybond filter with a 32P-labeled α-1,2-mannosidase-specific probe. Labeling of the probe was done using the High Prime kit (Roche). The DNA template for the labeling reaction consisted of a part of the gpdA promoter and was obtained through an EcoRI (Promega)/NheI (Biolabs) digest on plasmid pFGPDGLAT3-ManHDEL. Integration was also checked by PCR on 1 μg gDNA using Taq polymerase (MBI Fermentas). A gene-specific antisense primer hybridizing against the 3' region of the mannosidase gene (5'-CAACTCGTCGTGAGCAAGG-3') (SEQ ID NO:25), and a sense primer that hybridizes against the gpdA promoter region of the expression vector (5'-CCATATTTTCCTGCTCTCCC-3') (SEQ ID NO:26), were used for the amplification reaction. The PCR conditions were as follows: 30 cycli of 1 min. at 94° C., 1 min. at 60° C. and 2 min. at 72° C.
[0100]For the analysis of the GlcNAc-transferase I transformants, genomic DNA was digested overnight with 50 units BglII (Promega). After electrophoresis, the DNA was transferred to a Hybond N.sup.+ membrane (Amersham). Integration of the expression plasmid into the genome was checked, by hybridizing the Hybond filter with a 32P-labeled GlcNAc transferase I-specific probe. Labeling of the probe was done using the High Prime kit (Roche). The DNA template for the labeling reaction consisted of a part of the GlcNAc transferase I ORF and was obtained through a BglII/NcoI digest on plasmid pFGPDKrecoGnTI.
Construction of the S. cerevisiae Plasmids
[0101]pSCGALMFHIFNB2 is an IFNβ expression construct where the IFNβ coding sequence is placed under control of the GAL promoter (Demolder et al., 1994)
[0102]The 3' end of the ROT2 gene was isolated by PCR reaction using 5'TACGGGCCCGGGAAAAAAACGAAGTGATATC3' (SEQ ID NO:27) as sense primer and 5'CCTTGTCGAGGTGGGAAATGTCC3' (SEQ ID NO:28) as antisense primer. The PCR conditions used were 95° C. for 3 min; 94° C. for 1 min; 55° C. for 1 min; 72° C. for 1 min; 25 cycli; 72° C. for 10 min; cool down to 4° C. The resulting fragment was cloned into pCR2.1-TOPO (Invitrogen Co, Carlsbad, Calif., USA) to yield pCR2.1-TOPO3'ROT2.
[0103]pGAPADE1glsII was constructed as follows: the glucosidase II ORF of S. cerevisiae was amplified from the gDNA of strain INVSc (α leu2-3, 112 his3Δ1, trp1-289, ura3-52) (Invitrogen). gDNA was prepared from an overnight grown yeast culture in YPD at 30° C. DNA was prepared using the Nucleon Kit for extraction of yeast gDNA (Amersham). The sense primer for the PCR amplification hybridizes to the 5' part of the yeast ORF (including the ATG start coding) and contains a XhoI restriction site for easier downstream cloning work. The antisense primer hybridizes against the 3' part of the ORF (but not including the stop codon) and contains an ApaI site for easier downstream cloning. The sequence of both primers is as follows: sense primer ROT2(S): 5'-CCGCTCGAGATGGTCCTTTTGAAATGGCTC-3' (SEQ ID NO:23) and antisense primer ROT2(AS): 5'-CCGGGCCCAAAAATAACTTCCCAATCTTCAG-3' (SEQ ID NO:29). PCR was done via a touch-down strategy using LA TaKaRa (ImTec Diagnostics) on 200 ng gDNA, using 50 pmol of each primer. The amplification was obtained during 3 rounds of 94° C. for 30 sec.--98° C. for 2 sec.--65° C. for 30 sec.--70° C. for 10 min., followed by 3 similar PCR rounds, however this time with an annealing temperature of 60° C., followed by 30 similar PCR rounds, however this time using an annealing temperature of 55° C.
[0104]A fragment of the expected length of 2900 bp was obtained via this PCR strategy and was XhoI/ApaI ligated into a XhoI/ApaI opened pGAPZA (Invitrogen). The resulting vector was called pGAPZAglsII and carries the S. cerevisiae glucosidase II alpha subunit under the transcriptional control of the Pichia GAP promoter. pGAPZAglsII was cut with NsiI,T4/PinAI to isolate a fragment containing the GAP promoter and glsII ORF. The obtained fragment, was ligated into a SalI/PinAI opened pBLADE 1×' plasmid creating vector pGAPADE1glsII. Vector pBLADE 1×' was a kind gift from Dr Benjamin Glick (Department of Molecular Genetics and Cell Biology, University of Chicago, USA) (Sears et al., 1998)
[0105]pCR2.1-TOPO3'ROT2 was cut with SalI EcoRI and treasted with 1 μl T4 (Boehringer Mannheim) with 1 μl dXTP (10 mM) and 1 μl of appropriate buffer for 1 hr at 37° C. The resulting fragment plasmid was cloned into a T4 treated SalI cut pGAPADEIglsII to yield pGAPADE1glsII3'binv.
[0106]A 1222 bp SphI SnaBI URA3 gene fragment of S. cerevisiae was cloned into SphI Eco RV opened pGAPADE1glsII3'binv to give pKOROT2.
[0107]pGAPADE1glsII3'binv was used as template to introduce the T. reesei mutation in the S. cerevisiae glucosidase II gene. The mutagenesis was carried out using 5'GTAGGATCCTCGCAAAGCC3' (SEQ ID NO:30) as mutation sense primer and 5'GACAATTACATTGAGGAAAGATCCG3' (SEQ ID NO:31) as mutation antisense primer. The reaction mixture consisted of 80 μl H2O, 10 μl buffer with (NH4)2SO4--MgCl2, 6 μl MgCl2, 2 μl dXTP (10 mM), 1 μl mutation sense primer (100 pmol/μl), 1 μl mutation antisense primer (100 pmol/μl), 0.5 μl template DNA and 0.5 μl Taq DNA polymerase. The reaction conditions used were 95° C. for 2 min, 94° C. for 1 min, 54° C. for 1 min, 72° C. for 1 min, 24 cycles (from step 2), 72° C. for 10 min, cool down till 4° C.
[0108]The mutant fragment was reintroduced in pGAPADE1glsII3'binv as a BamHI XcmI fragment and the resulting plasmid was called pGAPADE1GLSIImut3'.
[0109]The T4 polymerase treated EcoO109I fragment, which contains a LEU2 ORF, of the plasmid YipUTYL was cloned into a T4 treated DraIII/XbaI cut pYX132 to yield pYX132LEU.
[0110]The vector pYX132 was purchased from Ingenius (R&D Systems Europe, Abingdon, UK). The vecor YipUTYL was taken form the LMBP plasmid collection (LMBP 3871).
[0111]pGAPADE1GLSIImut3' was cut with EcoRI and treated with T4 polymerase, and the GLSII mutant containing fragment was cloned into a cip treated SmaI opened pYX132LEUste. The resulting plasmid was called pYX132LEUglsIImut3'.
N-Glycan Analysis
[0112]Transformants were grown for 6 days at 30° C., in 100 ml shake flasks containing 50 ml minimal medium with glucose, lactose or cellulose as single carbon source (composition per liter: 20 g dextrose monohydrate or lactose or Solca Floc cellulose, 5 g (NH4)2SO4, 15 g KH2PO4, 0.3 g CaCl2, 0.3 g MgSO4 and mineral components). N-glycans of the total pool of secreted proteins were prepared according to Papac et al. (1998) from 1 ml of growth medium. The final glycan pellet was resuspended into 5 μl of bidest H2O. 1 μl of this glycan preparation was used for oligosaccharide analysis by DSA-FACE, as described recently (Callewaert, et al., 2001).
[0113]Mild acid hydrolysis of the N-glycans was performed on 1 μl of the prepared N-glycan mixture by incubation with 9 μl 10 mM HCl at 100° C. during 30 min. Before DSA-FACE analysis, the sample was dried and the pellet resuspended into 1 μl bidest H2O. In vitro α-1,2-mannosidase and β-N-Acetylglucosminidase digestions were done overnight at 37° C. on 1 μl of the prepared N-glycan mixture in 20 mM NaOAc pH 5.0. As enzyme source, in house produced T. Reesei α-1,2-mannosidase (Maras et al., 2000) and Jack Bean derived hexosaminidase (Glyko) were used. Before DSA-FACE analysis, the sample was dried and the pellet resuspended into 1 μl bidest H2O.
Analysis of Secreted Protein
Using Shake Flask Cultures:
[0114]T. Reesei RutC30 WT and transformant g14, expressing a full-size copy of the T. Reesei glucosidase II alpha subunit, were grown for 6 days at 30° C., in 100 ml shake flasks containing 50 ml of minimal medium with glucose as single carbon source (composition per litter: 20 g dextrose monohydrate, 5 g (NH4)2SO4, 15 g KH2PO4, 0.3 g CaCl2, 0.3 g MgSO4 and mineral components). After growth, the mycelium was separated from the medium and dried overnight at 50° C. Total extracellular protein of a fraction of the growth medium was TCA precipitated. The volume for the different samples taken for the precipitation of the total protein, was normalized against the dry weight of the mycelium. The precipitated proteins were resuspended in loading buffer and analyzed by SDS-PAGE. Gels were stained using coomassie brilliant blue (Sigma).
Using Steady-State Growth Conditions:
[0115]T. Reesei strains QM9414, Rut-C30 and its glucosidase II alpha subunit transformant Rut-C30-g31 were grown in steady-state/chemostat conditions. Briefly, the strains were grown at 28° C. with a dilution rate of 0.05 h-1. The culture medium consists of 8 g/l lactose, 3.75 g/1 KH2PO4, 5.7 g/l (NH4)2SO4, 0.17 g/l CaCl2.2H2O, 0.375 μl MgSO4.7H2O and 1 ml/l of a trace element solution consisting of 3.7 g/l CoCl2, 5 g/l FeSO4.7H2O, 1.4 g/l ZnSO4.7H2O and 1.6 g/l MnSO4.7H2O. The pH was kept constant at 5.5: adjustments were done automatically with 0.1 N KOH. Foaming was controlled by a mixture of polypropylene glycols. Samples of the chemostat culture were taken at regular time-intervals. Total cellulase activity was measured with para-Nitrophenyl-β-D-lactopyranoside as a substrate and compared to a standard curve of T. Reesei cellulases (Sigma). 1 unit releases 1 μmol op para-Nitrophenol per hour at 37° C. Total protein concentration was measured using the Bradford assay, with T. Reesei cellulases from Sigma as standard protein.
Analysis of the Transformants by Lectin Screening
[0116]Transformants were grown for 6 days at 30° C., in 100 ml shake flasks containing 50 ml minimal medium with glucose as single carbon source. 1 ml of growth medium was used to precipitate the secreted proteins with trichloroacetic acid. Proteins were separated by SDS-PAGE and blotted onto nitrocellulose membranes, using standard techniques (Sambrook et al., 1989).
[0117]The nitrocellulose membrane was blocked with TNT-buffer (50 mM Tris.HCl pH 7.5; 150 mM NaCl; 0.1% Tween-20) for 1 hour and washed briefly in lectin buffer (50 mM Tris.HCl pH 7.5; 150 mM NaCl; 0.05% Tween-20; 1 mM MgCl2; 1 mM CaCl2; 1 mM MnCl2). Afterwards, the membrane was incubated for 2 hours with a biotinylated Griffonia simplicifolia II lectin, which is specific for terminal GlcNAc (EY laboratories, Inc.). The lectin was diluted in lectin buffer according to the specifications of the provider. The membrane was washed twice (15 minutes in lectin buffer) and incubated for 1 hour in lectin buffer with steptavidin conjugated to peroxidase (Roche). After two wash steps (15 minutes in lectin buffer), the peroxidase was detected using the Renaissance® chemiluminescence kit (NEN® Life Science). Luminescent signals were captured either using the Lumi-Imager® F1 apparatus from Boehringer Mannheim or on an X-ray film.
IFNβ Western Blots
[0118]Western blots were carried out as described by Redlich and Grossberg (1989) and Grossberg et al., 1986.
[0119]IFNβ secretion was tested on a 15% polyacrylamide gel. The primary antibody as an anti-human IFNβ monoclonal antibody (Chemicon International, Temecula, Calif., USA). The secondary antibody was a goet anti-mouse HRP conjugated monoclonal anti-IgG1 antibody (Apovia). Visualization was carried out using a Western Lightning Chemiluminescence Reagent Plus kit (Perkin Elmer Life Sciences, Boston, Mass., USA)
Bio-Informatics
[0120]Conversion of nucleotide sequences into amino acid sequences was done using the Translate Tool at us.expasy.org/tools/#translate. Homology searches were done using the BLAST algorithm at www.ch.embnet.org/software/BottomBLAST.html (Altschul et al., 1990). Dual and multiple alignments were performed using the Clustal W algorithm (Thompson et al., 1994) at www.ebi.ac.uk/clustalw, resp. the Align program (GENESTREAM network server IGH, Montpellier FRANCE) at www2.igh.cnrs.fr/bin/align-guess.cgi (Pearson et al., 1997). General features of the protein (MW, pI, Amino acid composition, . . . ) were assessed using the ProtParam Tool at us.expasy.org/tools/protparam.html. The presence of a putative signal sequence was predicted using Signal P (version 1.1) at www.cbs.dtu.dk/services/SignalP. Prediction of the presence of transmembrane helices was done using the TMHMM (version 2.0) program at www.cbs.dtu.dk/services/TMHMM-2.0 or the HMMTOP (version 2.0) program (by G. E. Tusnady) at www.enzim.hu/hmmtop. All above-mentioned tools are either local or accessible via a link on the ExPASy (Expert Protein Analysis System) proteomics server from the Swiss Institute of Bioinformatics (SIB) (Appel et al., 1994).
Example 1
Cloning of the T. Reesei Glucosidase II Alpha Subunit Gene
[0121]cDNA Cloning of the Glucosidase II Alpha Subunit
[0122]Using the ClustalW algorithm website, an alignment was made between the amino acid sequences of the S. cerevisiae glucosidase II and the several known mammalian glucosidase II alpha subunits. Based on several homologous regions, three degenerate primers were designed to screen a cDNA library of the T. Reesei Rut-C30 strain (VTT Biotechnology). Amplification using sense primer 1, antisense primer 3 and the cDNA library as template DNA, resulted into a fragment of the expected size of 1170 bp (FIG. 3a). Nested PCR amplification including antisense primer 2, resulted into an extra DNA fragment with an expected length of about 970 bp (FIG. 3b). Both fragments were cloned in the TOPO-TA vector pCR2.1-TOPO (Invitrogen) for sequence analysis. By homology search, the obtained nucleotide sequences proved to be glucosidase II specific.
[0123]Based on this knowledge, cloning of the glucosidase II alpha subunit cDNA was started from the Rut-C30 cDNA library, using the technique of "cDNA cloning by PCR screening" (Takuma and Lodish, 1994). The PCR analysis was performed using sense primer 1 and antisense primer 3. Each PCR round (three in total) indicated that several wells within the microtiterplate contained at least one glucosidase II specific clone (FIG. 4). In the final PCR round each well contained a cell suspension of about 50 different cDNA clones. Two of these wells proved to be positive during the PCR screening. A dilution of the cell suspension of one of those wells was plated on solid Luria Bertani medium. About 200 colonies were streaked on filters for colony hybridization. Using a 32P labeled probe, we identified 7 positive clones (FIG. 5). DNA of the 7 clones was prepared and analyzed through a XhoI/EcoRI digestion. Fragments of about 1700 bp, 600 bp and 200 bp were obtained and proved to be glucosidase II specific either by southern hybridization (FIG. 6) or by sequence analysis after cloning into pUC19 or pBluescript II KS +/-. The completely cloned cDNA fragment consisted of 2290 bp. Homology analysis indicated that a substantial part of the 5' end of the ORF was missing.
Cloning of the 5' Coding Sequence of the Glucosidase II Alpha Subunit
[0124]Both an inverse PCR strategy and a 5'RACE strategy were initiated to clone the missing part. Through inverse PCR on Rut-C30 genomic DNA, a 1700 bp fragment was obtained (FIG. 7a) and cloned into the pCR2.1-TOPO vector. Partial sequence analysis indicated the presence of two fragments showing homology to the 5' part of the ORF of other known glucosidase II genes. The two glucosidase II homologous regions were separated from each other by a 60 bp sequence, containing features that are specific for intron sequences in filamentous fungi. The existence of an intron at the 5' end of the Trichoderma glucosidase II sequence was further confirmed by a 5' RACE strategy. Using the "First Choice®" RLM-RACE kit (Ambion) we obtained an 1138 bp fragment (FIG. 7b) missing the 60 bp intron sequence, but otherwise showing a 100% homology to the already cloned iPCR fragment (FIG. 7c).
Intron Analysis
[0125]To evaluate the intron-exon composition of the Trichoderma glucosidase II alpha subunit gene, a PCR was performed on genomic Rut-C30 DNA using 5' and 3' gene-specific primers. Amplification resulted into a fragment of about 3000 bp, which is close to the length of the coding cDNA. This indicated that only a few, rather small introns could be present. Alignment of the PCR fragment with the cloned cDNA showed that the 60 bp intron at the 5' terminus was the only intron present within the glucosidase II alpha subunit gene. The small size of this intron is consistent with sizes of most other characterized introns in filamentous fungi (May et al., 1987; Martinez-Blanco et al., 1993; Takaya et al., 1994). The intron follows the GT/AG rule for the 5' and 3' splice site (Mount, 1982). 13 nucleotides upstream of the 3' splice site, the intron contains a lariat sequence of the consensus CTRAC (with R=purine), which is characteristic for other fungal introns (Hiraoka et al., 1984; Orbach et al., 1986; May et al., 1987).
Frame-Shift Analysis
[0126]The DNA sequencing data of the 5' RACE fragment and the cloned cDNA sequence were put together, resulting in a 3621 bp fragment. Translation and BLAST analysis indicated the presence of an ORF showing homology to known glucosidase II alpha subunits. The glucosidase II ORF encodes a polypeptide of 807 amino acids. Contrary to the first 655 amino acids, the C-terminal 152 amino acids do not show any considerable sequence homology to other known glucosidase II alpha subunits. On top of that, the Trichoderma glucosidase II polypeptide sequence is significantly smaller compared to the yeast or mammalian homologue. This indicated the presence of a frame-shift within the cloned cDNA, resulting into a premature abrogation of translation. Indeed, computer analysis of the 3' 1500 bp of the 3621 bp fragment showed the presence of an out-of-frame sequence of 927 bp encoding a polypeptide of 309 amino acids, which shows high homology to the C-terminus of known glucosidase II alpha subunits.
[0127]Using two glucosidase II internal primers ROT2TR3_S and ROT2TR0_AS, a fragment of about 320 bp was amplified from the genomic DNA of T. Reesei Rut-C30 and QM9414. Based on the BLAST homology searches, the annealing sites of the two primers were chosen so that the amplified fragment should contain the site of the frame-shift. Sequence alignment of the two PCR fragments clearly indicates the presence of a frame-shift in the Rut-C30 genome, which was not found within the QM9414 genome: at position 1965 of the glucosidase II alpha subunit ORF a `T` is missing. As such, a premature stop codon, 459 nucleotides 3' of the position of the frame-shift, results in a truncated protein with 153 C-terminal amino acids that are not specific to the Trichoderma glucosidase II alpha subunit. This mutation within the Rut-C30 genome could very well explain the difference in glycosylation pattern when comparing the Rut-C30 strain with other T. Reesei strains.
General Features of the Cloned Gene
[0128]The knowledge of the nature of the mutation within the glucosidase II alpha subunit gene enabled us to put together some general data on the Trichoderma glucosidase II. A full-size non-mutant ORF of 2892 bp encodes a polypeptide of 964 amino acids, which is about the expected length based on data from other known glucosidase II alpha subunits. The protein has a calculated molecular weight of 109.858 Dalton and a theoretical pI of 5.6. Analysis using Signal P (version 1.1) indicated the presence of a putative eukaryotic signal sequence of 30 amino acids. A signal cleavage site was predicted after Leu29Ala30. Analysis of the mature polypeptide sequence with the TMHMM (version 2.0) and the HMMTOP (version 2.0) program did not reveal the presence of transmembrane helices. The polypeptide sequence also seems to lack any known ER-retention signal such as an HDEL tag. These data are in agreement with the general model for the glucosidase II protein: the alpha subunit is the catalytic partner of a heterodimer that is retained within the ER by interacting with the beta subunit, which carries an ER retention signal. The T. Reesei glucosidase II alpha subunit has the highest sequence homology (64.2% identity) to the Neurospora crassa counterpart, while the sequence identity with the Saccharomyces cerevisiae homologue is only 37.9%. On the other hand, sequence identity with Schizosaccharomyces pombe and higher eukaryotic organisms like pig, human and Arabidopsis thaliana is resp. 43.1, 40.9, 40.4 and 40.1%.
Example 2
Expression of a Fully Active Trichoderma Glucosidase II Alpha Subunit in the Rut-C30 Strain
Construction of a Trichoderma Glucosidase II Alpha Subunit Expression Plasmid and Transformation to Rut-C30
[0129]A T. Reesei expression vector encoding a functional variant of the Rut-C30 glucosidase II alpha subunit was prepared. In a first step, the frame-shift within the cloned cDNA fragment was repaired. In a next step, the 5' RACE fragment and the repaired cDNA were ligated to one another to obtain a full length ORF, encoding a full size and functional alpha subunit. In a last step, the ORF was placed under the transcriptional control of the constitutive gpdA promoter and the TrpC terminator resulting in the vector pFGPDglsIITreesei. Using a second strategy, a similar vector was created in which the 10 C-terminal codons of the ORF were replaced by the coding sequence of the Myc-tag, resulting into vector pFGPDglsIITreeseiMyc.
[0130]Both plasmids were transformed to T. Reesei Rut-C30 as described by Penttila et al (1987) using pAN7.1 as selection plasmid (Punt et al., 1987). Transformants were selected on their resistance to hygromycin. After two rounds of clone purification, single clones were obtained.
Analysis of the Transformants
[0131]The initial analysis of the transformants was based on the functionality of the expressed glucosidase II alpha subunit. For that, transformants were initially grown for 6 days in 50 ml glucose minimal medium, after which the N-glycan profile of the pool of total secreted protein was analyzed. N-glycans were prepared from 0.25 to 1 ml of growth medium as described by Papac and coworkers (1998) and analyzed by DSA-FACE (Callewaert et al, 2001a). The N-glycan profiles of the QM9414 strain, which does not carry monoglucosylated N-glycans (Garcia et al., 2001), and the RutC30 WT strain were analyzed in the same way and compared with that of the selected transformants.
[0132]Based on the published structural data of the most predominant oligosaccharides synthesized on secreted cellobiohydrolase I (De Bruyn et al, 1997, Maras et al., 2000), the profile of the RutC30 strain appeared relatively easy to interpret. An in vitro α-1,2-mannosidase digestion was used to characterize the peaks representing monoglucosylated high-mannose glycans. Since the α-1,3-linked glucose blocks the hydrolysis of the two underlying α-1,2-linked mannose residues, a maximum of two mannoses can be released from the glycan substrate, resulting in GlcMan7GlcNAc2. Mild acid hydrolysis, which hydrolyzes phosphodiester linkages, was used to characterize the peaks representing phosphorylated high-mannose carbohydrates. Release of the phosphate-coupled mannose results in a phosphomonoester glycan, which carries an extra negative charge and as such has a higher electrophoretic mobility. Peaks representing these glycans are shifted to the left side of the DSA-FACE profile. As such, using a combination of an α-1,2-mannosidase digestion and a mild acid hydrolysis, the most predominant peaks within the DSA-FACE glycan pattern of the RutC30 strain could be assigned to GlcMan7-8GlcNAc2 and their charged counterparts ManPGlcMan7-8GlcNAc2 (FIG. 8).
[0133]A similar analysis was performed on the glycan pattern of QM9414. Initially, the QM9414 DSA-FACE profile looks far more complex. However, comparison with the standard profile of RNase B indicates that a significant fraction of the glycan pool consists of Man5-9GlcNAc2. This was confirmed via an in vitro digestion with α-1,2-mannosidase. Moreover, mild acid hydrolysis of the carbohydrates indicates that most of the peaks at the left hand side of the Man5GlcNAc2 signal represent glycans containing one or more phosphodiester linkages. As such, the QM9414 glycan peaks could be assigned to neutral and phosphorylated high-mannose N-glycans. The distribution of the phosphorylated N-glycans is not severely changed after α-1,2-mannosidase digestion, since the phosphodiester linkages form a steric hindrance for the enzyme or block the access to underlying α-1,2-linked mannose residues (FIG. 8).
[0134]In a next step, the N-glycan profile of several hygromycin resistant transformants was analyzed. Only one of the analyzed transformants (g14) shows a clear difference in its N-glycan pattern, compared to the RutC30 WT strain. The g14 glycan pool looks more heterogeneous and closer examination indicates that it consists of a combination of the RutC30 and the QM9414 carbohydrate profile (FIG. 8). Especially at the left hand side of the Man5GlcNAc2 peak, a lot of new peaks emerge representing fast migrating oligosaccharides. Since most of them are susceptible to mild acid hydrolysis, we believe that they represent a structural diversity of phosphorylated high-mannose glycans, analogous to the situation in QM9414. In combination with these charged high-mannose N-glycans, some peaks representing neutral unglucosylated carbohydrates also emerge in the DSA-FACE profile. The presence of these structures was further confirmed by performing an in vitro α-1,2-mannosidase digestion. Comparison of the g14 with the RutC30 profile however, clearly indicates that still a significant amount of monoglucosylated glycans (neutral and charged GlcMan7-9GlcNAc2) is synthesized on the proteins of the transformed strain. Presumably, the amount of full-size glucosidase II is not sufficient to hydrolyze all α-1,3-linked glucose residues.
[0135]A number of hygromycin resistant transformants, including g14, and the WT RutC30 strain were analyzed on the genomic level. To clearly discriminate between endogenous mutant alpha subunit locus and the repaired cDNA from the integrated expression vector, genomic DNA was digested with KpnI/NheI (NheI cuts within the gpdA promoter of the expression vector) and analyzed via Southern blot using a glucosidase II specific probe. Using this strategy, two bands of approximately 5000 and 3400 bp were visualized for the g14 transformant, the latter one resulting from a random integration of the alpha subunit expression cassette into the T. Reesei genome. For all other transformants showing no change in their N-glycan profile, only the fragment of about 5000 bp was identified. This is identical to the band obtained for the untransformed RutC30 strain and as such can only result from a hybridization event to the endogenous locus encoding the truncated GIIα.
Effect of the Overexpression of the Glucosidase II Alpha Subunit on the Secretion Capacity of the Transformants
[0136]The T. Reesei RutC30 strain, which is a hypersecretor of cellulases, synthesizes unusual N-glycan structures on its secreted proteins. Thorough analysis indicated that most of the oligosaccharides carries α-1,3-glucose residues (Maras et al. 1997). These capping structures, which prevent further trimming of the glycans by α-1,2-mannosidase, are probably the result of an inefficient glucosidase II activity. Several events may cause this phenomenon: (1) the glucosidase II simply cannot cope the rich load of protein passing through the secretion pathway during cellulase-inducing conditions; (2) during the consecutive rounds of mutagenesis leading to this hypersecretor strain, one or more mutations have occurred within the glucosidase II gene or its transcriptional elements.
[0137]Surprisingly, we found that restoration of the glucosidase II activity affects the secretion capacity that is similar to that of the wild type strain. Transformant g14, where both glucosidase II forms are expressed, shows a secretion capacity that is significantly lower than that of the hypersecreting stain RutC30, clearly indicating that the glucosidase II plays an important role in the level of secretion.
Example 3
Expression of α-1,2-Mannosidase in Trichoderma
[0138]In order to localize most of the recombinant α-1,2-mannosidase to the ER compartment, where it can act on the substrate Man8GlcNAc2 to deliver a Man5GlcNAc2 structure, an HDEL-tag was added at the C-terminal end of the protein. By doing so the recombinant protein is recycled in a COP I depended manner from the Golgi apparatus to the ER due to binding to the HDEL-receptor.
[0139]An expression cassette containing the constitutive gpdA promoter, the prepro-signal sequence of the yeast α-mating factor for directing the protein into the secretory pathway, the HDEL-tagged α-1,2-mannosidase ORF and the trpC terminator was constructed. The construct was transformed using AmdS (acetamidase) as a selection marker. Transformants were selected for their ability to grow on minimal medium with acetamide as a sole nitrogen source. The transformants were submitted to several selection rounds in order to get single or "pure" clones.
[0140]To assess the functionality of the ER-localized α-1,2-mannosidase, the N-glycan profile of the total pool of secreted protein was analyzed. For this, transformants were grown in glucose containing minimal medium as described in materials and methods. N-glycans were released from 1 ml of growth medium through the method described by Papac et al., 1998. The results from the DSA-FACE analysis are depicted in FIG. 12. In total, 16 transformants were analyzed by DSA-FACE, of which 4 had the expected Man5GlcNAc2 glycan pattern. Evidence for the presence of the mannosidase expression plasmid in the genome of the transformants was obtained by Southern blotting/PCR analysis. Only a very small amount of the total glycan pool consists of Man6-9GlcNAc2, some of them carrying mannosephosphate. An additional in vitro digestion with purified recombinant α-1,2-mannosidase almost completely converts the remnant neutral high-mannose N-glycans to Man5GlcNAc2.
[0141]When grown in glucose minimal medium, the total amount of secreted cellulases is rather low. To evaluate the trimming capacity of the ER-localized α-1,2-mannosidase during cellulase inducing conditions, one of the positive transformants (transformant F4) was grown in minimal medium with 2% lactose, 5% lactose or SolcaFloc cellulose as C-source instead of 2% glucose. N-glycan analysis was as described in materials and methods. When comparing the N-glycan profiles from the different growth conditions, we found almost no difference. This suggests that the recombinant α-1,2-mannosidase is expressed in sufficient amounts to deal with a large flow of protein within the secretory pathway.
[0142]The results clearly indicate that the ER localization of a functional α-1,2-mannosidase enables the fungus to convert most of the ER high-mannose structure Man8-9GlcNAc2 to Man5GlcNAc2. By doing so, phosphomannosylation of the N-glycans is almost completely abolished. It seems that in the untransformed strain, the phosphomannosyltransferase and the α-1,2-mannosidase compete for the same high-mannose oligosaccharides. The obtained Man5GlcNAc2 structure is no substrate for the phosphomannosyltransferase, which is in accordance with data published for the S. cerevisiae Mnn6p transferase protein (Wang et al, 1997). This also suggests that the Trichoderma phosphomannosyltransferase activity is located somewhat further in the secretion pathway (medial to trans Golgi).
[0143]In conclusion, the obtained results indicate that by using this strategy, we can convert the fungal-type N-glycosylation pattern of T. Reesei to almost exclusively Man5GlcNAc2. Since this is the substrate for the GlcNAc transferase I, the key enzyme in the synthesis of complex N-glycans, new possibilities in creating a Trichoderma strain with a more human-like glycan profile can be explored.
Example 4
Expression of GlcNAc-Transferase in Trichoderma
[0144]For GlcNAc-transferase I to be localized to the Golgi compartment, where it can act on the Man5GlcNAc2 structure, two chimeras were created in the past between the C-terminal part of GlcNAc-transferase I and the N-terminal part of yeast Kre2, a Golgi localized mannosyltransferase (Lussier et al., 1995). The fusion positions were based on the fact that both proteins contain a putative coiled coil that might be important for localization and oligomerisation of the protein. Indeed, when the respective amino acid sequences were analyzed by the paircoil program (Berger et al., 1995), a coiled coil was predicted from amino acid 49 to 81 in GlcNAc-transferase I with a probability of 0.36 and from amino acid 54 to 99 in Kre2 with a probability of maximum 0.69 (see also FIG. 14). In addition, when analyzing the GlcNAc transferase I of other organisms, the probability of the presence of a coiled coil structure was even higher. Based on results obtained in Aspergillus niger, plasmid pFGPDKrecoGnTI encoding a chimer of the first 100 amino acids of Kre2 and the C-terminal part of GlcNAc-transferase I starting from amino acid 103 (as such having the coiled coil of Kre2), was preferred for the expression of recombinant human GlcNAc transferase I in T. Reesei.
[0145]The construct was cotransformed to the α-1,2-mannosidase expressing transformant F4 of strain QM9414. The Streptoalloteichus hindustanus phleomycin-binding protein expression cassette was used as selection marker. Transformants were selected based on their capacity to grow on minimal medium containing zeocin. The transformants were submitted to several selection rounds in order to get single or "pure" clones.
[0146]To identify functional GlcNAc-transferase I transformants a first screening round was performed using the Griffonia simplicifolia II lectin, which is specific for terminal GlcNAc residues. Several transformants were grown for 6 days on glucose minimal medium.
[0147]Transformants that score positive during the lectin screening were further analyzed by DSA-FACE. N-glycans were released from 1 ml of growth medium through the method described by Papac et al., 1998. Changes in the glycosylation profile that could indicate the functional expression of GlcNAc transferase I were investigated by in vitro digestion of with Jack Bean hexosaminidase (Glyko): strains of which the pattern returned to the Man5GlcNAc2 profile after the in vitro digestion, proof to be the desired glycosylation transformants.
Example 5
Effect of the Overexpression of the Glucosidase II Alpha Subunit on the Secretion Capacity of the Transformants
[0148]The T. Reesei RutC30 strain contains a frame-shift mutation in the glucosidase II alpha subunit gene, resulting in the production of a partially defective gene product. To restore the normal ER-processing of protein-linked N-glycans, this strain was transformed with the expression plasmid pFGPDglsIITreesei (FIG. 2), containing the full-size Trichoderma glucosidase II alpha subunit (GIIα) gene under the transcriptional control of the constitutive gpdA promoter. Transformation was done according to Penttila and coworkers (1987). Vector pAN7.1 (Punt et al., 1987) was co-transformed to enable selection of the transformants on hygromycin containing medium.
[0149]Several hygromycin-resistant clones were analyzed by DSA-FACE. As described in example 2, only one transformant (designated as g14) showed a severe difference in its N-glycan profile compared to the RutC30 untransformed strain. Southern analysis indicated that only g14 had randomly integrated the GIIα expression plasmid into its genome (FIG. 9).
[0150]Apart from the phenotype on the N-glycan level, also the secretion capacity of the g14 transformant seems to be affected. To analyze the effect on the production of extracellular proteins, several strains were grown on 50 ml minimal medium in 100 ml shake flasks. Incubations were performed for 6 to 7 days, at 30° C. and 150 rpm (rotations per minute). The minimal medium consists per liter of 20 g glucose, 5 g (NH4)2SO4, 15 g KH2PO4, 0.3 g CaCl2, 0.3 g MgSO4 and mineral components. Since all analyzed clones are derived from the RutC30 strain, their cellulose expression is not subject to carbon catabolyte repression due to the absence of a functional CRE1 (Ilmen, et al., 1996). Hence, a sufficient amount of extracellular hydrolases is synthesized to perform an SDS-PAGE analysis. The proteins are precipitated from the growth medium using TCA (trichloro-acetic acid), resuspended in 2× Laemmli loading buffer and analyzed via Gel electrophoresis (FIG. 15).
[0151]Interpretation of the observed SDS-PAGE profile indicates that the secretion capacity of the g14 transformant is reduced compared to the RutC30 WT and untransformed strains. The exercise was repeated several times to check the reproducibility of the obtained data. After growth on glucose minimal medium for 6 to 7 days, the level of the g14 secretion seems to be lower than that of the RutC30 strain.
Example 6
Construction of a Glucosidase II Knock Out in Saccharomyces cerevisiae
[0152]The strategy to construct the ROT2 knock out is summarized in FIG. 16. pKOROT2 is a vector comprising an integration cassette consisting of a S. cerevisiae URA3 expression cassette inserted between about 60 bp of the 5' end of the ROT2 ORF at one side and the 3' end untranslated region of this ORF at the other side. The plasmid pKOROT2 was digested with XhoI to release the integration cassette. Transformation of S. cerevisiae YA-72 with this cassette and selection on a URA.sup.- medium results in the selection of mutants in which the yeast glucosidase II gene has been replaced by the URA3 expression cassette. Transformants were tested using an upward primer in the URA3 gene and a downward primer in the 3' untranslated region. 3 out of 19 analyzed clones were showing the right insert. The positive clones were tested on their sugar profile using DSA-FACE analysis, and compared with a negative clone, with the parental strain and with the rot2 knock out mutant Y13369 and its parental strain BY4742 (FIG. 17). From the sugar profiles, it can indeed be concluded that the glucosidase II genes was inactivated in the transformants.
[0153]FIG. 16 indicates how the URA3 gene in the knock out further can be exchanged against a mutant glucosidase II gene, carrying the mutation that is found in the T reesei RutC30 glucosidase II.
Example 7
S. cerevisiae Strains with a Mutant Glucosidase II Show Increased Secretion
[0154]S. cerevisiae Y13369, as well as the parental strain BY4742 were transformed with the episomal plasmid pSCGALMFHIFNB2, carrying the human IFNβ gene preceded by the S. cerevisiae mating factor and under control of the GALL promoter. Transformants were selected on URAmedium. From both strain, 8 transformants were analyzed by Western Blotting. The yeast strains were precultivated for 48 hours in YPD, and the expression was induced for 48 hours in YPGal. The proteins, secreted in the medium were precipitated with TCA and separated using a 15% SDS-PAGE gel. Blotting was carried out by the semi-dry method, and the results are summarized in FIG. 18.
[0155]Although the results are not quantified, it is clear that in general the knockout mutants do secrete more IFNβ in the medium than the wild type strain.
[0156]To obtain more quantitative data, the experiment was repeated and the secretion was compared with the secretion of an IFNβ producing knock out complemented with a mutant glucosidase. This strain was obtained by transforming the IFNβ knock out strain with pYX132LEUGLSIImut3' and selection of SDC URA.sup.- LEU.sup.- medium.
[0157]8 individual transformants of each strain (Y133369 transformed with pSCGALMFHIFNB2, BY4742 transformed with pSCGALMFHIFNB2 and Y13369 transformed with both pSCGALMFHIFNB2 and pYX132LEUGLSIImut3') were grown in selective medium (SDC URA.sup.- or SDC URA.sup.- LEU.sup.-) for 18 hours. Then the cells were harvested and washed three times with water. The expression was induced by resuspending the cells in SDGal URA.sup.-, resp. SDGal URA.sup.- LEU.sup.- and cultivating them for another 24 hours. The cells were pelleted and the medium was collected. The supernatant of each of the 8 transformants was pooled. Two samples of the pooled supernatant, one of 0.5 ml and one of 1 ml was TCA precipitated. The proteins were separated using an SDS-PAGE gel, and blotted as described above. The results are summarized in FIG. 19 and Table 1. It is clear from these results that both the mutant glucosidase II and the knock out mutant show an increased secretion compared with the wild type.
TABLE-US-00001 TABLE 1 Quantification of the protein bands of FIG. 19, ad determined by Lumi Imager. A mut KO WT Mut 1 2.96 0.308 KO 0.338 1 0.104 WT 3.25 9.6 1 B Mut KO WT Mut 1 2.66 0.93 KO 0.376 1 0.35 WT 1.073 2.85 1 A: analysis of the 0.5 ml sample. B: analysis of the 1 ml sample. The values are expressed as relative intensity ratios.
REFERENCES
[0158]D'Alessio C, Fernandez F, Trombetta E S, Parodi A J. 1999. Genetic evidence for the heterodimeric structure of glucosidase II. The effect of disrupting the subunit-encoding genes on glycoprotein folding. J Biol Chem. 274:25899-905. [0159]Alonso J M, Santa-Cecilia A, Calvo P. 1993. Effect of bromoconduritol on glucosidase II from rat liver. A new kinetic model for the binding and hydrolysis of the substrate. Eur J Biochem. 215:37-42. [0160]Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. 1990. Basic local alignment search tool. J Mol Biol. 215:403-10. [0161]Appel R D, Bairoch A, Hochstrasser D F. A new generation of information retrieval tools for biologists: the example of the ExPASy WWW server. Trends Biochem Sci. 19:258-60. [0162]Arendt C W, Ostergaard H L. 1997. Identification of the CD45-associated 116-kDa and 80-kDa proteins as the alpha- and beta-subunits of alpha-glucosidase II. J Biol Chem. 272:13117-25. [0163]Berger B, Wilson D B, Wolf E, Tonchev T, Milla M, Kim P S. 1995. Predicting coiled coils by use of pairwise residue correlations. Proc Natl Acad Sci USA. 92:8259-63. [0164]Callewaert N, Geysens S, Molemans F, Contreras R. 2001. Ultrasensitive profiling and sequencing of N-linked oligosaccharides using standard DNA-sequencing equipment. Glycobiology, 11:275-81. [0165]Callewaert N, Laroy W, Cadirgi H, Geysens S, Saelens X, Min Jou W, Contreras R. 2001b. Use of HDEL-tagged T. Reesei mannosyl oligosaccharide 1,2-alpha-D-mannosidase for N-glycan engineering in Pichia pastoris. FEBS Lett. 503:173-8. [0166]Camirand A, Heysen A, Grondin B, Herscovics A. 1991. Glycoprotein biosynthesis in Saccharomyces cerevisiae. Isolation and characterization of the gene encoding a specific processing alpha-mannosidase. J Biol Chem. 266:15120-7. [0167]Casadaban M J, Cohen S N. 1980. Analysis of gene control signals by DNA fusion and cloning in Escherichia coli. J Mol Biol. 138:179-207. [0168]Chiba Y, Suzuki M, Yoshida S, Yoshida A, Ikenaga H, Takeuchi M, Jigami Y, Ichishima E. 1998. Production of human compatible high mannose-type (Man5GlcNAc2) sugar chains in Saccharomyces cerevisiae. J Biol Chem. 273:26298-304. [0169]Contreras R, Carrez D, Kinghom J R, van den Hondel C A, Fiers W. 1991. Efficient KEX2-like processing of a glucoamylase-interleukin-6 fusion protein by Aspergillus nidulans and secretion of mature interleukin-6. Biotechnology (NY) 9:378-81. [0170]De Bruyn A, Maras M, Schraml J, Herdewijn P, Contreras R. 1997. NMR evidence for a novel asparagine-linked oligosaccharide on cellobiohydrolase I from T. Reesei RUTC 30. FEBS Lett. 405:111-3. [0171]de Groot M J, Bundock P, Hooykaas P J, Beijersbergen A G. 1998. Agrobacterium tumefaciens-mediated transformation of filamentous fungi. Nat Biotechnol. 16:839-42. [0172]Demolder J, Fiers W, Contreras R. 1994. Human interferon-beta, expressed in Saccharomyces cerevisiae, is predominantly directed to the vacuoles. Influence of modified co-expression of secretion factors and chaperones. J Biotechnol 32:179-89. [0173]Eades C J, Gilbert A M, Goodman C D, Hintz W E. 1998. Identification and analysis of a class 2 alpha-mannosidase from Aspergillus nidulans. Glycobiology, 8:17-33. [0174]Flura T, Brada D, Ziak M, Roth J. 1997. Expression of a cDNA encoding the glucose trimming enzyme glucosidase II in CHO cells and molecular characterization of the enzyme deficiency in a mutant mouse lymphoma cell line. Glycobiology, 7:617-24. [0175]Freeze H H. 1985. Interaction of Dictyostelium discoideum lysosomal enzymes with the mammalian phosphomannosyl receptor. The importance of oligosaccharides which contain phosphodiesters. J Biol Chem. 260:8857-64. [0176]Fukada T, Iida K, Kioka N, Sakai H, Komano T. 1994. Cloning of a cDNA encoding N-acetylglucosaminyltransferase I from rat liver and analysis of its expression in rat tissues. Biosci. Biotechnol. Biochem. 58:200-201. [0177]Garcia R, Cremata J A, Quintero O, Montesino R, Benkestock K, Stahlberg J. (2001) Characterization of protein glycoforms with N-linked neutral and phosphorylated oligosaccharides: studies on the glycosylation of endoglucanase 1 (Cel7B) from T. Reesei. Biotechnol Appl Biochem 33 (Pt2):141-152 [0178]Goldman G, Van Montagu M, Herrera-Estrella A. 1990. Transformation of Trichoderma harzianum by high-voltage electric pulse. Curr Genet. 17: 1169-1174. [0179]Grossberg, S E et al. (1986) in Manual of clinical immunology Rose, N R, Friedman, H R and Fahey, J L (eds.). ASM Press, Washington, D.C. 1986., pp 259-299. [0180]Herscovics A, Schneikert J, Athanassiadis A, Moremen K W. 1994. Isolation of a mouse Golgi mannosidase cDNA, a member of a gene family conserved from yeast to mammals. J. Biol. Chem. 269: 9864-9871. [0181]Hiraoka Y, Toda T, Yanagida M. 1984. The NDA3 gene of fission yeast encodes beta-tubulin: a cold-sensitive nda3 mutation reversibly blocks spindle formation and chromosome movement in mitosis. Cell, 39(2 Pt 1):349-58. [0182]Hynes M J, Corrick C M, King J A. 1983. Isolation of genomic clones containing the amdS gene of Aspergillus nidulans and their use in the analysis of structural and regulatory mutations. Mol Cell Biol. 3:1430-1439. [0183]Ilmen M, Thrane C, Penttila, M. (1996) The glucose repressor gene cre1 of Trichoderma: isolation and expression of a full-length and a truncated mutant form. Mol Gen Genet 251: 451-560. [0184]Kornfeld R, Kornfeld S. 1985. Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem. 54:631-64. [0185]Lal A, Schutzbach J S, Forsee W T, Neame P J, Moremen K W. 1994. Isolation and expression of murine and rabbit cDNAs encoding an alpha 1,2-mannosidase involved in the processing of asparagine-linked oligosaccharides. J. Biol. Chem. 269: 9872-9881. [0186]Lorito M, Hayes C K, Di Pietro A, Harman G E. 1993. Biolistic transformation of Trichoderma harzianum and Gliocladium virens using plasmid and genomic DNA. Curr Genet, 24:349-56. [0187]Lussier M, Sdicu A M, Ketela T, Bussey H. 1995. Localization and targeting of the Saccharomyces cerevisiae Kre2p/Mnt1p alpha 1,2-mannosyltransferase to a medial-Golgi compartment. J Cell Biol. 131:913-27. [0188]Maras M, De Bruyn A, Schraml J, Herdewijn P, Claeyssens M, Fiers W, Contreras R. 1997. Structural characterization of N-linked oligosaccharides from cellobiohydrolase I secreted by the filamentous fungus T. Reesei RUTC 30. Eur J Biochem. 245:617-25. [0189]Maras M, van Die I, Contreras R, van den Hondel C A. 1999. Filamentous fungi as production organisms for glycoproteins of bio-medical interest. Glycoconj J. 16:99-107 [0190]Maras M, Callewaert N, Piens K, Claeyssens M, Martinet W, Dewaele S, Contreras H, Dewerte I, Penttila M, Contreras R. 2000. Molecular cloning and enzymatic characterization of a Trichoderma reesei 1,2-alpha-D-mannosidase. J Biotechnol. 77(2-3):255-63. [0191]Martinet W, Maras M, Saelens X, Min Jou W, Contreras R. 1998. Modification of the protein glycosylation pathway in the methylotrophic yeast Pichia pastoris. Biotechnology Letters 20: 1171-1177. [0192]Martinez-Blanco H, Orejas M, Reglero A, Luengo J M, Penalva M A. 1993. Characterization of the gene encoding acetyl-CoA synthetase in Penicillium chrysogenum: conservation of intron position in plectomycetes. Gene, 130:265-70. [0193]Mattern I E, Punt P J, van den Hondel C A. 1988. Fungal Gen Newslett. 35, 25. [0194]May G S, Morris N R. 1987. The unique histone H2A gene of Aspergillus nidulans contains three introns. Gene, 58:59-66. [0195]Mount S M. 1982. A catalogue of splice junction sequences. Nucleic Acids Res. 10:459-72. [0196]Odani T, Shimma Y, Tanaka A, Jigami Y. 1996. Cloning and analysis of the MNN4 gene required for phosphorylation of N-linked oligosaccharides in Saccharomyces cerevisiae. Glycobiology, 6:805-10. [0197]Orbach M J, Porro E B, Yanofsky C. 1986. Cloning and characterization of the gene for beta-tubulin from a benomyl-resistant mutant of Neurospora crassa and its use as a dominant selectable marker. Mol Cell Biol. 6:2452-61. [0198]Papac D I, Briggs J B, Chin E T, Jones A J. 1998. A high-throughput microscale method to release N-linked oligosaccharides from glycoproteins for matrix-assisted laser desorption/ionization time-of-flight mass spectrometric analysis. Glycobiology, 8:445-54. [0199]Pearson, W. R., Wood, T., Zhang, Z., and Miller, W. 1997. Comparison of DNA sequences with protein sequences. Genomics 46: 24-36. [0200]Pelham H R. 1988. Evidence that luminal ER proteins are sorted from secreted proteins in a post-ER compartment. EMBO J. 7: 913-918. [0201]Penttila M, Nevalainen H, Ratto M, Salminen E, Knowles J. 1987. A versatile transformation system for the cellulolytic filamentous fungus Trichoderma reesei. Gene, 61:155-64. [0202]Punt P J, Oliver R P, Dingemanse M A, Pouwels P H, van den Hondel C A. 1987. Transformation of Aspergillus based on the hygromycin B resistance marker from Escherichia coli. Gene, 56:117-24. [0203]Punt P J, van Biezen N, Conesa A, Albers A, Mangnus J, van den Hondel C. 2002. Filamentous fungi as cell factories for heterologous protein production. Trends Biotechnol. 20:200-6. [0204]Redlich, P N and Grossberg, S E. 1989. Analysis of antigenic domains on natural and recombinant human IFN-beta by the inhibition of biologic activities with monoclonal antibodies. J. Immunol 143: 1887-93. [0205]Saloheimo A, Henrissat B, Hoffren A M, Teleman O, Penttila M. 1994. A novel, small endoglucanase gene, egl5, from Trichoderma reesei isolated by expression in yeast. Mol Microbiol, 13:219-28. [0206]Sambrook J, Fritsch E F, Maniatis T. (1989) Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y. [0207]Sarkar M, Hull E, Nishikawa Y, Simpson R. J, Moritz R. L, Dunn R, Schachter H. 1991. Molecular cloning and expression of cDNA encoding the enzyme that controls conversion of high-mannose to hybrid and complex N-glycans: UDP-N-acetylglucosamine: alpha-3-D-mannoside beta-1,2-N-acetylglucosaminyltransferase I. Proc. Natl. Acad. Sci. U.S.A. 88:234-238. [0208]Schachter H. 1991. Organization and localization to chromosome 5 of the human UDP-N-acetylglucosamine:alpha-3-D-mannoside beta-1,2-N-acetylglucosaminyltransferase I gene. Biochem. Biophys. Res. Commun. 176:608-615. [0209]Sears I B, O'Connor J, Rossanese O W and Glick BS. 1998. A versatile set of vectors for constitutive and regulated gene expression in Pichia pastoris. Yeast vol. 14, pg 783-790. [0210]Takaya N, Yanai K, Horiuchi H, Ohta A, Takagi M. 1994. Cloning and characterization of two 3-phosphoglycerate kinase genes of Rhizopus niveus and heterologous gene expression using their promoters. Curr Genet. 25:524-30. [0211]Takumi T, Lodish H F. 1994. Rapid cDNA cloning by PCR screening. Biotechniques, 17:443-444. [0212]Thompson J D, Higgins D G, Gibson T J. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-80. [0213]Tremblay L O, Campbell Dyke N, Herscovics A. 1998. Molecular cloning, chromosomal mapping and tissue-specific expression of a novel human alpha1,2-mannosidase gene involved in N-glycan maturation. Glycobiology 8: 585-595. [0214]Trombetta E S, Simons J F, Helenius A. 1996. Endoplasmic reticulum glucosidase II is composed of a catalytic subunit, conserved from yeast to mammals, and a tightly bound noncatalytic HDEL-containing subunit. J Biol Chem. 271(44):27509-16. [0215]Van Petegem F, Contreras H, Contreras R, Van Beeumen J. 2001. Trichoderma reesei alpha-1,2-mannosidase: structural basis for the cleavage of four consecutive mannose residues. J Mol Biol. 312:157-65. [0216]Wang X H, Nakayama K, Shimma Y, Tanaka A, Jigami Y. 1997. MNN6, a member of the KRE2/MNT1 family, is the gene for mannosylphosphate transfer in Saccharomyces cerevisiae. J Biol Chem. 272:18117-24.
Sequence CWU
1
3412670DNAHomo sapiensCDS(1)..(2670)misc_featureGnTI 1atg ctg aag aag cag
tct gca ggg ctt gtg ctg tgg ggc gct atc ctc 48Met Leu Lys Lys Gln
Ser Ala Gly Leu Val Leu Trp Gly Ala Ile Leu1 5
10 15ttt gtg gcc tgg aat gcc ctg ctg ctc ctc ttc
ttc tgg acg cgc cca 96Phe Val Ala Trp Asn Ala Leu Leu Leu Leu Phe
Phe Trp Thr Arg Pro 20 25
30gca cct ggc agg cca ccc tca gtc agc gct ctc gat ggc gac ccc gcc
144Ala Pro Gly Arg Pro Pro Ser Val Ser Ala Leu Asp Gly Asp Pro Ala35
40 45agc ctc acc cgg gaa gtg att cgc ctg gcc
caa gac gcc gag gtg gag 192Ser Leu Thr Arg Glu Val Ile Arg Leu Ala
Gln Asp Ala Glu Val Glu50 55 60ctg gag
cgg cag cgt ggg ctg ctg cag cag atc ggg gat gcc ctg tcg 240Leu Glu
Arg Gln Arg Gly Leu Leu Gln Gln Ile Gly Asp Ala Leu Ser65
70 75 80agc cag cgg ggg agg gtg ccc
acc gcg gcc cct ccc gcc cag ccg cgt 288Ser Gln Arg Gly Arg Val Pro
Thr Ala Ala Pro Pro Ala Gln Pro Arg 85 90
95gtg cct gtg acc ccc gcg ccg gcg gtg att ccc atc ctg gtc atc
gcc 336Val Pro Val Thr Pro Ala Pro Ala Val Ile Pro Ile Leu Val Ile
Ala 100 105 110tgt gac cgc agc act gtt
cgg cgc tgc ctg gac aag ctg ctg cat tat 384Cys Asp Arg Ser Thr Val
Arg Arg Cys Leu Asp Lys Leu Leu His Tyr115 120
125cgg ccc tcg gct gag ctc ttc ccc atc atc gtt agc cag gac tgc ggg
432Arg Pro Ser Ala Glu Leu Phe Pro Ile Ile Val Ser Gln Asp Cys Gly130
135 140cac gag gag acg gcc cag gcc atc gcc
tcc tac ggc agc gcg gtc acg 480His Glu Glu Thr Ala Gln Ala Ile Ala
Ser Tyr Gly Ser Ala Val Thr145 150 155
160cac atc cgg cag ccc gac ctg agc agc att gcg gtg ccg ccg
gac cac 528His Ile Arg Gln Pro Asp Leu Ser Ser Ile Ala Val Pro Pro
Asp His 165 170 175cgc aag ttc cag
ggc tac tac aag atc gcg cgc cac tac cgc tgg gcg 576Arg Lys Phe Gln
Gly Tyr Tyr Lys Ile Ala Arg His Tyr Arg Trp Ala 180
185 190ctg ggc cag gtc ttc cgg cag ttt cgc ttc ccc gcg
gcc gtg gtg gtg 624Leu Gly Gln Val Phe Arg Gln Phe Arg Phe Pro Ala
Ala Val Val Val195 200 205gag gat gac ctg
gag gtg gcc ccg gac ttc ttc gag tac ttt cgg gcc 672Glu Asp Asp Leu
Glu Val Ala Pro Asp Phe Phe Glu Tyr Phe Arg Ala210 215
220acc tat ccg ctg ctg aag gcc gac ccc tcc ctg tgg tgc gtc
tcg gcc 720Thr Tyr Pro Leu Leu Lys Ala Asp Pro Ser Leu Trp Cys Val
Ser Ala225 230 235 240tgg
aat gac aac ggc aag gag cag atg gtg gac gcc agc agg cct gag 768Trp
Asn Asp Asn Gly Lys Glu Gln Met Val Asp Ala Ser Arg Pro Glu 245
250 255ctg ctc tac cgc acc gac ttt ttc cct
ggc ctg ggc tgg ctg ctg ttg 816Leu Leu Tyr Arg Thr Asp Phe Phe Pro
Gly Leu Gly Trp Leu Leu Leu 260 265
270gcc gag ctc tgg gct gag ctg gag ccc aag tgg cca aag gcc ttc tgg
864Ala Glu Leu Trp Ala Glu Leu Glu Pro Lys Trp Pro Lys Ala Phe Trp275
280 285gac gac tgg atg cgg cgg ccg gag cag
cgg cag ggg cgg gcc tgc ata 912Asp Asp Trp Met Arg Arg Pro Glu Gln
Arg Gln Gly Arg Ala Cys Ile290 295 300cgc
cct gag atc tca aga acg atg acc ttt ggc cgc aag ggt gtg agc 960Arg
Pro Glu Ile Ser Arg Thr Met Thr Phe Gly Arg Lys Gly Val Ser305
310 315 320cac ggg cag ttc ttt gac
cag cac ctc aag ttt atc aag ctg aac cag 1008His Gly Gln Phe Phe Asp
Gln His Leu Lys Phe Ile Lys Leu Asn Gln 325 330
335cag ttt gtg cac ttc acc cag ctg gac ctg tct tac ctg cag
cgg gag 1056Gln Phe Val His Phe Thr Gln Leu Asp Leu Ser Tyr Leu Gln
Arg Glu 340 345 350gcc tat gac cga gat
ttc ctc gcc cgc gtc tac ggt gct ccc cag ctg 1104Ala Tyr Asp Arg Asp
Phe Leu Ala Arg Val Tyr Gly Ala Pro Gln Leu355 360
365cag gtg gag aaa gtg agg acc aat gac cgg aag gag ctg ggg gag
gtg 1152Gln Val Glu Lys Val Arg Thr Asn Asp Arg Lys Glu Leu Gly Glu
Val370 375 380cgg gtg cag tat acg ggc agg
gac agc ttc aag gct ttc gcc aag gct 1200Arg Val Gln Tyr Thr Gly Arg
Asp Ser Phe Lys Ala Phe Ala Lys Ala385 390
395 400ctg ggt gtc atg gat gac ctt aag tcg ggg gtt ccg
aga gct ggc tac 1248Leu Gly Val Met Asp Asp Leu Lys Ser Gly Val Pro
Arg Ala Gly Tyr 405 410 415cgg ggt
att gtc acc ttc cag ttc cgg ggc cgc cgt gtc cac ctg gcg 1296Arg Gly
Ile Val Thr Phe Gln Phe Arg Gly Arg Arg Val His Leu Ala 420
425 430ccc cca ccg acg tgg gag ggc tat gat cct agc
tgg aat atg ctg aag 1344Pro Pro Pro Thr Trp Glu Gly Tyr Asp Pro Ser
Trp Asn Met Leu Lys435 440 445aag cag tct
gca ggg ctt gtg ctg tgg ggc gct atc ctc ttt gtg gcc 1392Lys Gln Ser
Ala Gly Leu Val Leu Trp Gly Ala Ile Leu Phe Val Ala450
455 460tgg aat gcc ctg ctg ctc ctc ttc ttc tgg acg cgc
cca gca cct ggc 1440Trp Asn Ala Leu Leu Leu Leu Phe Phe Trp Thr Arg
Pro Ala Pro Gly465 470 475
480agg cca ccc tca gtc agc gct ctc gat ggc gac ccc gcc agc ctc acc
1488Arg Pro Pro Ser Val Ser Ala Leu Asp Gly Asp Pro Ala Ser Leu Thr
485 490 495cgg gaa gtg att cgc ctg gcc
caa gac gcc gag gtg gag ctg gag cgg 1536Arg Glu Val Ile Arg Leu Ala
Gln Asp Ala Glu Val Glu Leu Glu Arg 500 505
510cag cgt ggg ctg ctg cag cag atc ggg gat gcc ctg tcg agc cag cgg
1584Gln Arg Gly Leu Leu Gln Gln Ile Gly Asp Ala Leu Ser Ser Gln Arg515
520 525ggg agg gtg ccc acc gcg gcc cct ccc
gcc cag ccg cgt gtg cct gtg 1632Gly Arg Val Pro Thr Ala Ala Pro Pro
Ala Gln Pro Arg Val Pro Val530 535 540acc
ccc gcg ccg gcg gtg att ccc atc ctg gtc atc gcc tgt gac cgc 1680Thr
Pro Ala Pro Ala Val Ile Pro Ile Leu Val Ile Ala Cys Asp Arg545
550 555 560agc act gtt cgg cgc tgc
ctg gac aag ctg ctg cat tat cgg ccc tcg 1728Ser Thr Val Arg Arg Cys
Leu Asp Lys Leu Leu His Tyr Arg Pro Ser 565 570
575gct gag ctc ttc ccc atc atc gtt agc cag gac tgc ggg cac
gag gag 1776Ala Glu Leu Phe Pro Ile Ile Val Ser Gln Asp Cys Gly His
Glu Glu 580 585 590acg gcc cag gcc atc
gcc tcc tac ggc agc gcg gtc acg cac atc cgg 1824Thr Ala Gln Ala Ile
Ala Ser Tyr Gly Ser Ala Val Thr His Ile Arg595 600
605cag ccc gac ctg agc agc att gcg gtg ccg ccg gac cac cgc aag
ttc 1872Gln Pro Asp Leu Ser Ser Ile Ala Val Pro Pro Asp His Arg Lys
Phe610 615 620cag ggc tac tac aag atc gcg
cgc cac tac cgc tgg gcg ctg ggc cag 1920Gln Gly Tyr Tyr Lys Ile Ala
Arg His Tyr Arg Trp Ala Leu Gly Gln625 630
635 640gtc ttc cgg cag ttt cgc ttc ccc gcg gcc gtg gtg
gtg gag gat gac 1968Val Phe Arg Gln Phe Arg Phe Pro Ala Ala Val Val
Val Glu Asp Asp 645 650 655ctg gag
gtg gcc ccg gac ttc ttc gag tac ttt cgg gcc acc tat ccg 2016Leu Glu
Val Ala Pro Asp Phe Phe Glu Tyr Phe Arg Ala Thr Tyr Pro 660
665 670ctg ctg aag gcc gac ccc tcc ctg tgg tgc gtc
tcg gcc tgg aat gac 2064Leu Leu Lys Ala Asp Pro Ser Leu Trp Cys Val
Ser Ala Trp Asn Asp675 680 685aac ggc aag
gag cag atg gtg gac gcc agc agg cct gag ctg ctc tac 2112Asn Gly Lys
Glu Gln Met Val Asp Ala Ser Arg Pro Glu Leu Leu Tyr690
695 700cgc acc gac ttt ttc cct ggc ctg ggc tgg ctg ctg
ttg gcc gag ctc 2160Arg Thr Asp Phe Phe Pro Gly Leu Gly Trp Leu Leu
Leu Ala Glu Leu705 710 715
720tgg gct gag ctg gag ccc aag tgg cca aag gcc ttc tgg gac gac tgg
2208Trp Ala Glu Leu Glu Pro Lys Trp Pro Lys Ala Phe Trp Asp Asp Trp
725 730 735atg cgg cgg ccg gag cag cgg
cag ggg cgg gcc tgc ata cgc cct gag 2256Met Arg Arg Pro Glu Gln Arg
Gln Gly Arg Ala Cys Ile Arg Pro Glu 740 745
750atc tca aga acg atg acc ttt ggc cgc aag ggt gtg agc cac ggg cag
2304Ile Ser Arg Thr Met Thr Phe Gly Arg Lys Gly Val Ser His Gly Gln755
760 765ttc ttt gac cag cac ctc aag ttt atc
aag ctg aac cag cag ttt gtg 2352Phe Phe Asp Gln His Leu Lys Phe Ile
Lys Leu Asn Gln Gln Phe Val770 775 780cac
ttc acc cag ctg gac ctg tct tac ctg cag cgg gag gcc tat gac 2400His
Phe Thr Gln Leu Asp Leu Ser Tyr Leu Gln Arg Glu Ala Tyr Asp785
790 795 800cga gat ttc ctc gcc cgc
gtc tac ggt gct ccc cag ctg cag gtg gag 2448Arg Asp Phe Leu Ala Arg
Val Tyr Gly Ala Pro Gln Leu Gln Val Glu 805 810
815aaa gtg agg acc aat gac cgg aag gag ctg ggg gag gtg cgg
gtg cag 2496Lys Val Arg Thr Asn Asp Arg Lys Glu Leu Gly Glu Val Arg
Val Gln 820 825 830tat acg ggc agg gac
agc ttc aag gct ttc gcc aag gct ctg ggt gtc 2544Tyr Thr Gly Arg Asp
Ser Phe Lys Ala Phe Ala Lys Ala Leu Gly Val835 840
845atg gat gac ctt aag tcg ggg gtt ccg aga gct ggc tac cgg ggt
att 2592Met Asp Asp Leu Lys Ser Gly Val Pro Arg Ala Gly Tyr Arg Gly
Ile850 855 860gtc acc ttc cag ttc cgg ggc
cgc cgt gtc cac ctg gcg ccc cca ccg 2640Val Thr Phe Gln Phe Arg Gly
Arg Arg Val His Leu Ala Pro Pro Pro865 870
875 880acg tgg gag ggc tat gat cct agc tgg aat
2670Thr Trp Glu Gly Tyr Asp Pro Ser Trp Asn 885
8902890PRTHomo sapiensmisc_featureGnTI 2Met Leu Lys Lys
Gln Ser Ala Gly Leu Val Leu Trp Gly Ala Ile Leu1 5
10 15Phe Val Ala Trp Asn Ala Leu Leu Leu Leu
Phe Phe Trp Thr Arg Pro 20 25
30Ala Pro Gly Arg Pro Pro Ser Val Ser Ala Leu Asp Gly Asp Pro Ala35
40 45Ser Leu Thr Arg Glu Val Ile Arg Leu Ala
Gln Asp Ala Glu Val Glu50 55 60Leu Glu
Arg Gln Arg Gly Leu Leu Gln Gln Ile Gly Asp Ala Leu Ser65
70 75 80Ser Gln Arg Gly Arg Val Pro
Thr Ala Ala Pro Pro Ala Gln Pro Arg 85 90
95Val Pro Val Thr Pro Ala Pro Ala Val Ile Pro Ile Leu Val Ile
Ala 100 105 110Cys Asp Arg Ser Thr Val
Arg Arg Cys Leu Asp Lys Leu Leu His Tyr115 120
125Arg Pro Ser Ala Glu Leu Phe Pro Ile Ile Val Ser Gln Asp Cys
Gly130 135 140His Glu Glu Thr Ala Gln Ala
Ile Ala Ser Tyr Gly Ser Ala Val Thr145 150
155 160His Ile Arg Gln Pro Asp Leu Ser Ser Ile Ala Val
Pro Pro Asp His 165 170 175Arg Lys
Phe Gln Gly Tyr Tyr Lys Ile Ala Arg His Tyr Arg Trp Ala 180
185 190Leu Gly Gln Val Phe Arg Gln Phe Arg Phe Pro
Ala Ala Val Val Val195 200 205Glu Asp Asp
Leu Glu Val Ala Pro Asp Phe Phe Glu Tyr Phe Arg Ala210
215 220Thr Tyr Pro Leu Leu Lys Ala Asp Pro Ser Leu Trp
Cys Val Ser Ala225 230 235
240Trp Asn Asp Asn Gly Lys Glu Gln Met Val Asp Ala Ser Arg Pro Glu
245 250 255Leu Leu Tyr Arg Thr Asp Phe
Phe Pro Gly Leu Gly Trp Leu Leu Leu 260 265
270Ala Glu Leu Trp Ala Glu Leu Glu Pro Lys Trp Pro Lys Ala Phe
Trp275 280 285Asp Asp Trp Met Arg Arg Pro
Glu Gln Arg Gln Gly Arg Ala Cys Ile290 295
300Arg Pro Glu Ile Ser Arg Thr Met Thr Phe Gly Arg Lys Gly Val Ser305
310 315 320His Gly Gln Phe
Phe Asp Gln His Leu Lys Phe Ile Lys Leu Asn Gln 325
330 335Gln Phe Val His Phe Thr Gln Leu Asp Leu Ser Tyr
Leu Gln Arg Glu 340 345 350Ala Tyr Asp
Arg Asp Phe Leu Ala Arg Val Tyr Gly Ala Pro Gln Leu355
360 365Gln Val Glu Lys Val Arg Thr Asn Asp Arg Lys Glu
Leu Gly Glu Val370 375 380Arg Val Gln Tyr
Thr Gly Arg Asp Ser Phe Lys Ala Phe Ala Lys Ala385 390
395 400Leu Gly Val Met Asp Asp Leu Lys Ser
Gly Val Pro Arg Ala Gly Tyr 405 410
415Arg Gly Ile Val Thr Phe Gln Phe Arg Gly Arg Arg Val His Leu Ala
420 425 430Pro Pro Pro Thr Trp Glu Gly
Tyr Asp Pro Ser Trp Asn Met Leu Lys435 440
445Lys Gln Ser Ala Gly Leu Val Leu Trp Gly Ala Ile Leu Phe Val Ala450
455 460Trp Asn Ala Leu Leu Leu Leu Phe Phe
Trp Thr Arg Pro Ala Pro Gly465 470 475
480Arg Pro Pro Ser Val Ser Ala Leu Asp Gly Asp Pro Ala Ser
Leu Thr 485 490 495Arg Glu Val Ile
Arg Leu Ala Gln Asp Ala Glu Val Glu Leu Glu Arg 500
505 510Gln Arg Gly Leu Leu Gln Gln Ile Gly Asp Ala Leu
Ser Ser Gln Arg515 520 525Gly Arg Val Pro
Thr Ala Ala Pro Pro Ala Gln Pro Arg Val Pro Val530 535
540Thr Pro Ala Pro Ala Val Ile Pro Ile Leu Val Ile Ala Cys
Asp Arg545 550 555 560Ser
Thr Val Arg Arg Cys Leu Asp Lys Leu Leu His Tyr Arg Pro Ser 565
570 575Ala Glu Leu Phe Pro Ile Ile Val Ser
Gln Asp Cys Gly His Glu Glu 580 585
590Thr Ala Gln Ala Ile Ala Ser Tyr Gly Ser Ala Val Thr His Ile Arg595
600 605Gln Pro Asp Leu Ser Ser Ile Ala Val
Pro Pro Asp His Arg Lys Phe610 615 620Gln
Gly Tyr Tyr Lys Ile Ala Arg His Tyr Arg Trp Ala Leu Gly Gln625
630 635 640Val Phe Arg Gln Phe Arg
Phe Pro Ala Ala Val Val Val Glu Asp Asp 645 650
655Leu Glu Val Ala Pro Asp Phe Phe Glu Tyr Phe Arg Ala Thr
Tyr Pro 660 665 670Leu Leu Lys Ala Asp
Pro Ser Leu Trp Cys Val Ser Ala Trp Asn Asp675 680
685Asn Gly Lys Glu Gln Met Val Asp Ala Ser Arg Pro Glu Leu Leu
Tyr690 695 700Arg Thr Asp Phe Phe Pro Gly
Leu Gly Trp Leu Leu Leu Ala Glu Leu705 710
715 720Trp Ala Glu Leu Glu Pro Lys Trp Pro Lys Ala Phe
Trp Asp Asp Trp 725 730 735Met Arg
Arg Pro Glu Gln Arg Gln Gly Arg Ala Cys Ile Arg Pro Glu 740
745 750Ile Ser Arg Thr Met Thr Phe Gly Arg Lys Gly
Val Ser His Gly Gln755 760 765Phe Phe Asp
Gln His Leu Lys Phe Ile Lys Leu Asn Gln Gln Phe Val770
775 780His Phe Thr Gln Leu Asp Leu Ser Tyr Leu Gln Arg
Glu Ala Tyr Asp785 790 795
800Arg Asp Phe Leu Ala Arg Val Tyr Gly Ala Pro Gln Leu Gln Val Glu
805 810 815Lys Val Arg Thr Asn Asp Arg
Lys Glu Leu Gly Glu Val Arg Val Gln 820 825
830Tyr Thr Gly Arg Asp Ser Phe Lys Ala Phe Ala Lys Ala Leu Gly
Val835 840 845Met Asp Asp Leu Lys Ser Gly
Val Pro Arg Ala Gly Tyr Arg Gly Ile850 855
860Val Thr Phe Gln Phe Arg Gly Arg Arg Val His Leu Ala Pro Pro Pro865
870 875 880Thr Trp Glu Gly
Tyr Asp Pro Ser Trp Asn 885
89032208DNASaccharomyces cerevisiaemisc_featureScKre2 cDNA 3atg gcc ctc
ttt ctc agt aag aga ctg ttg aga ttt acc gtc att gca 48Met Ala Leu
Phe Leu Ser Lys Arg Leu Leu Arg Phe Thr Val Ile Ala1 5
10 15ggt gcg gtt att gtt ctc ctc cta aca
ttg aat tcc aac agt aga act 96Gly Ala Val Ile Val Leu Leu Leu Thr
Leu Asn Ser Asn Ser Arg Thr 20 25
30cag caa tat att ccg agt tcc atc tcc gct gca ttt gat ttt acc tca
144Gln Gln Tyr Ile Pro Ser Ser Ile Ser Ala Ala Phe Asp Phe Thr Ser35
40 45gga tct ata tcc cct gaa caa caa gtc
atc tct gag gaa aat gat gct 192Gly Ser Ile Ser Pro Glu Gln Gln Val
Ile Ser Glu Glu Asn Asp Ala50 55 60aaa
aaa tta gag caa agt gct ctg aat tca gag gca agc gaa gac tcc 240Lys
Lys Leu Glu Gln Ser Ala Leu Asn Ser Glu Ala Ser Glu Asp Ser65
70 75 80gaa gcc atg gat gaa gaa
tcc aag gct ctg aaa gct gcc gct gaa aag 288Glu Ala Met Asp Glu Glu
Ser Lys Ala Leu Lys Ala Ala Ala Glu Lys 85 90
95gca gat gcc ccg atc gac act aaa aca acc atg gat tat atc
act cca 336Ala Asp Ala Pro Ile Asp Thr Lys Thr Thr Met Asp Tyr Ile
Thr Pro 100 105 110tct ttt gct aac aaa
gct ggt aag cca aaa gct tgt tac gtc act ttg 384Ser Phe Ala Asn Lys
Ala Gly Lys Pro Lys Ala Cys Tyr Val Thr Leu115 120
125gtg aga aac aag gag ttg aaa ggt ttg cta agc tcc att aaa tat
gtg 432Val Arg Asn Lys Glu Leu Lys Gly Leu Leu Ser Ser Ile Lys Tyr
Val130 135 140gaa aac aaa att aac aag aaa
ttc cca tat cct tgg gtt ttc cta aac 480Glu Asn Lys Ile Asn Lys Lys
Phe Pro Tyr Pro Trp Val Phe Leu Asn145 150
155 160gat gaa cct ttt act gaa gaa ttc aag gaa gca gtc
acc aaa gct gtt 528Asp Glu Pro Phe Thr Glu Glu Phe Lys Glu Ala Val
Thr Lys Ala Val 165 170 175tct tcc
gaa gtt aag ttt ggt att ttg ccc aag gaa cat tgg tca tat 576Ser Ser
Glu Val Lys Phe Gly Ile Leu Pro Lys Glu His Trp Ser Tyr 180
185 190cct gaa tgg att aat caa acc aag gct gct gaa
att cgt gca gat gct 624Pro Glu Trp Ile Asn Gln Thr Lys Ala Ala Glu
Ile Arg Ala Asp Ala195 200 205gcc acc aaa
tac ata tac ggt ggc tcc gaa tct tat aga cac atg tgt 672Ala Thr Lys
Tyr Ile Tyr Gly Gly Ser Glu Ser Tyr Arg His Met Cys210
215 220cgt tac caa tct ggg ttt ttc tgg aga cat gaa tta
tta gaa gag tac 720Arg Tyr Gln Ser Gly Phe Phe Trp Arg His Glu Leu
Leu Glu Glu Tyr225 230 235
240gat tgg tac tgg cgt gtg gaa cca gac atc aag tta tac tgt gat att
768Asp Trp Tyr Trp Arg Val Glu Pro Asp Ile Lys Leu Tyr Cys Asp Ile
245 250 255aat tac gac gtt ttt aag tgg
atg caa gaa aac gaa aaa gtt tac ggc 816Asn Tyr Asp Val Phe Lys Trp
Met Gln Glu Asn Glu Lys Val Tyr Gly 260 265
270ttt acc gtt tct att cat gaa tat gaa gtg acg atc cca aca cta tgg
864Phe Thr Val Ser Ile His Glu Tyr Glu Val Thr Ile Pro Thr Leu Trp275
280 285caa acg tcc atg gat ttc atc aaa aag
aac ccc gaa tac tta gat gaa 912Gln Thr Ser Met Asp Phe Ile Lys Lys
Asn Pro Glu Tyr Leu Asp Glu290 295 300aac
aac ctg atg agt ttt ctt tcg aac gat aat ggt aaa aca tac aat 960Asn
Asn Leu Met Ser Phe Leu Ser Asn Asp Asn Gly Lys Thr Tyr Asn305
310 315 320ctg tgc cat ttc tgg tca
aac ttt gaa att gca aac ttg aat ttg tgg 1008Leu Cys His Phe Trp Ser
Asn Phe Glu Ile Ala Asn Leu Asn Leu Trp 325 330
335agg tca cca gcc tac aga gag tat ttt gac act ttg gat cat
caa ggt 1056Arg Ser Pro Ala Tyr Arg Glu Tyr Phe Asp Thr Leu Asp His
Gln Gly 340 345 350gga ttt ttc tac gaa
aga tgg ggc gat gct ccc gtt cat tct att gct 1104Gly Phe Phe Tyr Glu
Arg Trp Gly Asp Ala Pro Val His Ser Ile Ala355 360
365gct gct ttg ttt ttg cca aag gat aaa atc cat tat ttt tca gac
att 1152Ala Ala Leu Phe Leu Pro Lys Asp Lys Ile His Tyr Phe Ser Asp
Ile370 375 380ggt tac cat cat cca cct tat
gat aac tgc cca ttg gac aag gag gtc 1200Gly Tyr His His Pro Pro Tyr
Asp Asn Cys Pro Leu Asp Lys Glu Val385 390
395 400tat aac agt aac aac tgt gaa tgt gac caa ggt aat
gat ttc act ttc 1248Tyr Asn Ser Asn Asn Cys Glu Cys Asp Gln Gly Asn
Asp Phe Thr Phe 405 410 415caa ggt
tac tct tgt ggt aag gaa tat tat gat gct caa ggg ttg gta 1296Gln Gly
Tyr Ser Cys Gly Lys Glu Tyr Tyr Asp Ala Gln Gly Leu Val 420
425 430aag cca aaa aac tgg aaa aaa ttc cgt gag tag
aaatcttgga acatactgtt 1349Lys Pro Lys Asn Trp Lys Lys Phe Arg Glu435
440tctttgtttt gactttatac tttctattta tattttattt ttataactgg
ttaagtacac 1409ataggactgc gtatcaaaca tataagtgag gcaatccaca ttttttttaa
agattcgaat 1469atttttattc tcattagcgt attccgagaa tagttcgaaa aaatataagg
tatatcaaga 1529gtttttacaa gtgagaggaa agaggaataa gctataagca acaaaagcgt
aaaaaaatta 1589gctgaagaca tagaactatg gatgttctca aagaggtgtt gtcactagac
caagataaat 1649ttgaccagct gaaggaaacg agccgagata aaacaaatga aacggatgat
ccttttgaaa 1709actatttgaa ggattgtaaa tttaaagcgc cttcaaacaa agatcagtca
ccatttgcta 1769aacttaaatc attacaggaa actcattcta acaatgaagc ggctattaat
ataattattc 1829ctcaattgat tgattactta accgaattca ctaataggtt atcaaattac
acacaagatt 1889tagacttcat taaaaaaaag tccaatgaat tacagtcatt gctcgaatac
aactccacta 1949aactggcaca tatctctcct atggttaatg atttgatgat tcctcctgaa
ctcattgatg 2009acatcattaa agggaagatc aatgaaagct ggcaggataa tataacattc
atagcagata 2069aagaagaaat ttataacaag tataggtcca ataatctcga tcaagacaac
aaggacgcag 2129aaaattcagc aatgctagca ccaaaggatt ttgataagtt atgtcaactc
ctggacatcc 2189taaaaaatgt tattctaga
22084442PRTSaccharomyces cerevisiaemisc_featureScKre2 cDNA
4Met Ala Leu Phe Leu Ser Lys Arg Leu Leu Arg Phe Thr Val Ile Ala1
5 10 15Gly Ala Val Ile Val Leu
Leu Leu Thr Leu Asn Ser Asn Ser Arg Thr 20 25
30Gln Gln Tyr Ile Pro Ser Ser Ile Ser Ala Ala Phe Asp
Phe Thr Ser35 40 45Gly Ser Ile Ser Pro
Glu Gln Gln Val Ile Ser Glu Glu Asn Asp Ala50 55
60Lys Lys Leu Glu Gln Ser Ala Leu Asn Ser Glu Ala Ser Glu Asp
Ser65 70 75 80Glu Ala
Met Asp Glu Glu Ser Lys Ala Leu Lys Ala Ala Ala Glu Lys 85
90 95Ala Asp Ala Pro Ile Asp Thr Lys Thr Thr
Met Asp Tyr Ile Thr Pro 100 105 110Ser
Phe Ala Asn Lys Ala Gly Lys Pro Lys Ala Cys Tyr Val Thr Leu115
120 125Val Arg Asn Lys Glu Leu Lys Gly Leu Leu Ser
Ser Ile Lys Tyr Val130 135 140Glu Asn Lys
Ile Asn Lys Lys Phe Pro Tyr Pro Trp Val Phe Leu Asn145
150 155 160Asp Glu Pro Phe Thr Glu Glu
Phe Lys Glu Ala Val Thr Lys Ala Val 165 170
175Ser Ser Glu Val Lys Phe Gly Ile Leu Pro Lys Glu His Trp Ser
Tyr 180 185 190Pro Glu Trp Ile Asn Gln
Thr Lys Ala Ala Glu Ile Arg Ala Asp Ala195 200
205Ala Thr Lys Tyr Ile Tyr Gly Gly Ser Glu Ser Tyr Arg His Met
Cys210 215 220Arg Tyr Gln Ser Gly Phe Phe
Trp Arg His Glu Leu Leu Glu Glu Tyr225 230
235 240Asp Trp Tyr Trp Arg Val Glu Pro Asp Ile Lys Leu
Tyr Cys Asp Ile 245 250 255Asn Tyr
Asp Val Phe Lys Trp Met Gln Glu Asn Glu Lys Val Tyr Gly 260
265 270Phe Thr Val Ser Ile His Glu Tyr Glu Val Thr
Ile Pro Thr Leu Trp275 280 285Gln Thr Ser
Met Asp Phe Ile Lys Lys Asn Pro Glu Tyr Leu Asp Glu290
295 300Asn Asn Leu Met Ser Phe Leu Ser Asn Asp Asn Gly
Lys Thr Tyr Asn305 310 315
320Leu Cys His Phe Trp Ser Asn Phe Glu Ile Ala Asn Leu Asn Leu Trp
325 330 335Arg Ser Pro Ala Tyr Arg Glu
Tyr Phe Asp Thr Leu Asp His Gln Gly 340 345
350Gly Phe Phe Tyr Glu Arg Trp Gly Asp Ala Pro Val His Ser Ile
Ala355 360 365Ala Ala Leu Phe Leu Pro Lys
Asp Lys Ile His Tyr Phe Ser Asp Ile370 375
380Gly Tyr His His Pro Pro Tyr Asp Asn Cys Pro Leu Asp Lys Glu Val385
390 395 400Tyr Asn Ser Asn
Asn Cys Glu Cys Asp Gln Gly Asn Asp Phe Thr Phe 405
410 415Gln Gly Tyr Ser Cys Gly Lys Glu Tyr Tyr Asp Ala
Gln Gly Leu Val 420 425 430Lys Pro Lys
Asn Trp Lys Lys Phe Arg Glu435 4405100PRTSaccharomyces
cerevisiaeMISC_FEATUREScKre2 Golgi localization signal 5Met Ala Leu Phe
Leu Ser Lys Arg Leu Leu Arg Phe Thr Val Ile Ala1 5
10 15Gly Ala Val Ile Val Leu Leu Leu Thr Leu
Asn Ser Asn Ser Arg Thr 20 25
30Gln Gln Tyr Ile Pro Ser Ser Ile Ser Ala Ala Phe Asp Phe Thr Ser35
40 45Gly Ser Ile Ser Pro Glu Gln Gln Val Ile
Ser Glu Glu Asn Asp Ala50 55 60Lys Lys
Leu Glu Gln Ser Ala Leu Asn Ser Glu Ala Ser Glu Asp Ser65
70 75 80Glu Ala Met Asp Glu Glu Ser
Lys Ala Leu Lys Ala Ala Ala Glu Lys 85 90
95Ala Asp Ala Pro 100623DNAArtificial Sequenceprimer 1
6gtntatggna tnccngagca tgc
23721DNAArtificial Sequenceprimer 2 7gngcgtgngc ncngaagaan g
21822DNAArtificial Sequenceprimer 3
8tgnnnnccng cgaagaangc nc
22920DNAArtificial SequencePCR primer ROT2TR4_AS 9gttaaacgtt tcgtcccacc
201019DNAArtificial
SequencePCR primer ROT2TR1_S 10ggctccatcc ctttcatgc
191120DNAArtificial SequencePCR primer
ROT2TR-RLMRACE 11gatatactcg aagacgtcgg
201218DNAArtificial Sequencesense primer 12atgaggtcga
cgatgggg
181317DNAArtificial Sequenceantisense primer 13agccagcttg atgctcc
171422DNAArtificial
Sequenceprimer ROT2TR3_S 14tatctctggt ttcccgttct cg
221521DNAArtificial Sequenceprimer ROT2TR0_AS
15ctggtcatca atcgccaagc c
211620DNAArtificial Sequenceprimer ROT2TR2_S 16atcaatgagc aactcctggc
201731DNAArtificial
Sequencesense primer used to induce the silent mutation 17ccatgtgaag
gcccgggttg gggatgactg g
311831DNAArtificial Sequenceantisense primer used to induce the silent
mutation 18ccagtcatcc ccaacccggg ccttcacatg g
311927DNAArtificial Sequencesense primer 19gaattcccgc
ggtacgtaat tatgagg
272027DNAArtificial Sequenceantisense primer 20gtcgacctca taattacgta
ccgcggg 272127DNAArtificial
Sequenceprimer ROT2ScNco_S 21cttgccatgg tccttttgaa atggctc
272233DNAArtificial Sequenceprimer
ROT2ScMycHind_AS 22cccaagcttc tacagatcct cttctgagat gag
332330DNAArtificial Sequenceprimer ROT2Sc_S 23ccgctcgaga
tggtcctttt gaaatggctc
302430DNAArtificial Sequenceprimer ROT2Sc_AS 24ccgggcccaa aaataacttc
ccaatcttca 302519DNAArtificial
Sequencegene specific antisense primer 25caactcgtcg tgagcaagg
192620DNAArtificial Sequencesense
primer 26ccatattttc ctgctctccc
202731DNAArtificial SequenceSense primer ROT2 gene 27tacgggcccg
ggaaaaaaac gaagtgatat c
312823DNAArtificial SequenceAntisense primer for ROT2 gene 28ccttgtcgag
gtgggaaatg tcc
232931DNAArtificial SequenceAntisense primer ROT2(AS) 29ccgggcccaa
aaataacttc ccaatcttca g
313019DNAArtificial SequenceMutation sense primer 30gtaggatcct cgcaaagcc
193125DNAArtificial
SequenceMutation antisense primer 31gacaattaca ttgaggaaag atccg
2532300DNAArtificial SequenceInverse PCR
fragment 32atgaggtcga cgatggggct gtcctggaag tggacggcac tcttcagcct
tttaggtgcc 60attctgtgcc tgatcgggcc tgcctgtatg catcatgaac acatcgcggc
tttggaagca 120tcttgctgac attgaaacct tctagtggcc gtcaaggaac acgagttcaa
aaagtgccac 180caggccggct tctgcaaccg aaaccgtgca ttggccgacc ttgcggcttc
ccagagctcg 240acctgggtgt ctccctacaa ggctgtcttc gaatctccct cgttggaaga
cggaaagatt 30033240DNAArtificial Sequence5' Race fragment 33atgaggtcga
cgatggggct gtcctggaag tggacggcac tcttcagcct tttaggtgcc 60attctgtgcc
tgatcgggcc tgccttggcc gtcaaggaac acgagttcaa aaagtgccac 120caggccggct
tctgcaaccg aaaccgtgca ttggccgacc ttgcggcttc ccagagctcg 180acctgggtgt
ctccctacaa ggctgtcttc gaatctccct cgttggaaga cggaaagatt
240344PRTSaccharomyces cerevisiaeHDEL ER-retention sequence 34His Asp Glu
Leu1
User Contributions:
comments("1"); ?> comment_form("1"); ?>Inventors list |
Agents list |
Assignees list |
List by place |
Classification tree browser |
Top 100 Inventors |
Top 100 Agents |
Top 100 Assignees |
Usenet FAQ Index |
Documents |
Other FAQs |
User Contributions:
Comment about this patent or add new information about this topic:
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
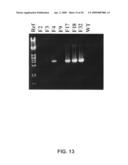 |  |
 |
| Similar patent applications: | |
| Date | Title |
|---|---|
| 2013-12-26 | Protein secretion |
| 2012-01-12 | Protein particles |
| 2010-02-04 | Fusion partner cells |
| 2011-12-01 | Hepatocyte lineage cells |
| 2014-07-17 | Protein extraction |
| New patent applications in this class: | |
| Date | Title |
|---|---|
| 2022-05-05 | Engineered cd47 extracellular domain for bioconjugation |
| 2019-05-16 | High cell density anaerobic fermentation for protein expression |
| 2019-05-16 | Polynucleotide encoding fusion of anchoring motif and dehalogenase, host cell including the polynucleotide, and use thereof |
| 2019-05-16 | Cell culture method, medium, and medium kit |
| 2018-01-25 | Protein expression strains |
| New patent applications from these inventors: | |
| Date | Title |
|---|---|
| 2014-12-25 | Filamentous fungi having reduced udp-galactofuranose content |
| 2014-06-05 | Protein glycosylation modification in methylotrophic yeast |
| 2011-05-05 | Filamentous fungi having reduced udp-galactofuranose content |
| 2011-02-17 | Novel vaccine against trypanosoma cruzi infection |
| 2010-10-21 | Protein glycosylation modification in methylotrophic yeast |
| Top Inventors for class "Chemistry: molecular biology and microbiology" | |
| Rank | Inventor's name |
|---|---|
| 1 | Marshall Medoff |
| 2 | Anthony P. Burgard |
| 3 | Mark J. Burk |
| 4 | Robin E. Osterhout |
| 5 | Rangarajan Sampath |