Patent application title: AMPLIFICATION OF HUMAN MDM2 GENE IN HUMAN TUMORS
Inventors:
Marilee Burrell (Cambridge, MA, US)
David E. Hill (Arlington, MA, US)
Kenneth W. Kinzler (Baltimore, MD, US)
Kenneth W. Kinzler (Baltimore, MD, US)
Bert Vogelstein (Baltimore, MD, US)
Assignees:
THE JOHNS HOPKINS UNIVERSITY
IPC8 Class: AC12Q168FI
USPC Class:
435 6
Class name: Chemistry: molecular biology and microbiology measuring or testing process involving enzymes or micro-organisms; composition or test strip therefore; processes of forming such composition or test strip involving nucleic acid
Publication date: 2008-12-11
Patent application number: 20080305490
Inventors list |
Agents list |
Assignees list |
List by place |
Classification tree browser |
Top 100 Inventors |
Top 100 Agents |
Top 100 Assignees |
Usenet FAQ Index |
Documents |
Other FAQs |
Patent application title: AMPLIFICATION OF HUMAN MDM2 GENE IN HUMAN TUMORS
Inventors:
Bert Vogelstein
Marilee Burrell
David E. Hill
Kenneth W. Kinzler
Agents:
BANNER & WITCOFF, LTD.
Assignees:
The Johns Hopkins University
Origin: WASHINGTON, DC US
IPC8 Class: AC12Q168FI
USPC Class:
435 6
Abstract:
A human gene has been discovered which is genetically altered in human
tumor cells. The genetic alteration is gene amplification and leads to a
corresponding increase in gene products. Detecting that the gene,
designated hMDM2, has become amplified or detecting increased expression
of gene products is diagnostic of tumorigenesis. Human MD2 protein binds
to human p53 and allows the cell to escape from p53-regulated growth.Claims:
1. A method of diagnosing a neoplastic tissue in a human
comprising:detecting amplification of human MDM2 gene or elevated
expression of a human MDM2 gene product in a tissue or body fluid
isolated from a human, wherein amplification of the human MDM2 gene or
elevated expression of human MDM2 gene product provides a diagnosis of
neoplasia or the potential for neoplastic development.
2. The method of claim 1 wherein gene amplification is detected.
3. The method of claim 1 wherein elevated expression of a gene product is detected, said gene product being mRNA.
4. The method of claim 1 wherein elevated expression of a gene product is detected, said gene product being human MDM2 protein.
5. The method of claim 3 wherein said mRNA is detected by Northern blot analysis by hybridizing mRNA from said tissue to a human MDM2 nucleotide probe.
6. The method of claim 5 wherein the human MDM2 nucleotide probe comprises nucleotides 1-2372 of human MDM2, as shown in FIG. 1, or fragments thereof consisting of at least 14 contiguous nucleotides.
7. The method of claim 4 wherein human MDM2 protein is detected by Western Blot analysis by reacting human MDM2 proteins with antibodies which are immunospecific for MDM2 protein.
8. The method of claim 2 wherein the gene amplification is detected using polymerase chain reaction.
9. The method of claim 2 wherein amplification of the human MDM2 gene is detected by Southern blot analysis wherein the human MDM2 gene is hybridized with a nucleotide probe which is complementary to hMDM2 DNA.
10. The method of claim 2 wherein gene amplification is determined by comparing the copy number of hMDM2 in the tissue to the copy number of hMDM2 in a normal tissue of the human.
11. The method of claim 3 wherein elevated expression of mRNA is determined by comparing the amount of hMDM2 mRNA in the tissue to the amount of hMDM2 mRNA in a normal tissue of the human.
12. The method of claim 4 wherein elevated expression of hMDM2 protein is determined by comparing the amount of hMDM2 protein in the tissue to the amount of hMDM2 protein in a normal tissue of the human.
13. The method of claim 2 wherein gene amplification is detected when at least 3-fold more hMDM-2 DNA is observed in the tissue relative to a control sample comprising a normal tissue.
14. The method of claim 3 wherein elevated expression is detected when at least 3-fold more hMDM-2 mRNA is observed in the tissue relative to a control sample comprising a normal tissue.
15. The method of claim 4 wherein elevated expression is detected when at least 3-fold more hMDM2 protein is observed in the tissue relative to a control sample comprising a normal tissue.
16. The method of claim 1 wherein the neoplasia is a sarcoma.
17. The method of claim 16 wherein the sarcoma is a liposarcoma, malignant fibrous histiocytoma, or osteosarcoma.
18. A cDNA molecule comprising nucleotides 1 to 2372, as shown in FIG. 1, or fragments thereof, consisting of at least 14 contiguous nucleotides.
19. The cDNA molecule of claim 18 comprising the coding sequence of human MDM2.
20. Human MDM2 protein substantially free of other human proteins.
21. A preparation of antibodies specifically immunoreactive with human MDM2 protein.
22. The preparation of claim 21 wherein the antibodies are monoclonal antibodies.
23. A nucleotide probe comprising a sequence of at least 10 nucleotides which are complementary to nucleotides 1-2372 of human MDM2 gene, as shown in FIG. 1.
24. A kit for detecting the amplification of a human MDM2 gene in a human tissue or body fluid sample comprising: a nucleic acid probe capable of hybridizing to said human MDM2 gene under conditions of high stringency, and instructions for determining said amplification.
25. A kit for detecting elevated expression of a human MDM2 mRNA in a human tissue or body fluid sample comprising: a nucleic acid probe capable of hybridizing to said mRNA, and written instructions for determining elevated expression of mRNA.
26. A kit for detecting elevated expression of a human MDM2 protein in a human tissue or body fluid sample comprising MDM2 protein-specific antibodies and written instructions for determining elevated expression of human MDM2 protein.
27. A method of treating a neoplastic cell or a cell having neoplastic potential, comprising:administering to a cell a therapeutically effective amount of an inhibitory compound which interferes with the expression of human MDM2 gene.
28. The method of claim 27 wherein expression of the human MDM2 gene is inhibited by administering antisense oligonucleotides.
29. The method of claim 27 wherein expression of the human MDM2 gene is inhibited by administering triple-strand forming oligonucleotides which interact with DNA.
30. A method for identifying compounds which interfere with the binding of human MDM-2 to human p53, comprising:binding a predetermined quantity of a first human protein which is detectably labelled to a second human protein;adding a compound to be tested for its capacity to inhibit binding of said first and second proteins to each other;determining the quantity of the first human protein which is displaced from or prevented from binding to the second human protein;wherein the first human protein is MDM-2 and the second human protein is p53 or the first human protein is p53 and the second human protein is MDM-2.
31. The method of claim 30 wherein one of said two human proteins is fixed to a solid support.
32. The method of claim 30 wherein an antibody specifically immunoreactive with said second human protein is used to separate first human protein bound from unbound first human protein.
33. A method for inhibiting the growth of tumor cells which contain a human MDM2 gene amplification, comprising:administering a polypeptide to tumor cells which contain a human MDM2 gene amplification, said polypeptide consisting essentially of a portion of p53, said portion comprising amino acids 13-41 of p53, said polypeptide being capable of binding to human MDM2.
34. The method of claim 33 wherein said polypeptide comprises amino acids 1-41 of p53.
35. The method of claim 33 wherein said polypeptide comprises amino acids 13-57 of p53.
36. The method of claim 33 wherein said polypeptide comprises amino acids 1-50 of p53.
37. A method for inhibiting the growth of tumor cells which contain a human MDM2 gene amplification, comprising:administering to tumor cells which contain a human MDM2 gene amplification a DNA molecule which expresses a polypeptide consisting essentially of a portion of p53, said portion comprising amino acids 13-41 of p53, said polypeptide being capable of binding to human MDM2.
38. The method of claim 37 wherein said polypeptide comprises amino acids 1-41 of p53.
39. The method of claim 37 wherein said polypeptide comprises amino acids 13-57 of p53.
40. The method of claim 37 wherein said polypeptide comprises amino acids 1-50 of p53.
41. A polypeptide consisting essentially of a portion of p53, said portion comprising amino acids 13-41 of p53, said polypeptide capable of binding to human MDM2.
42. The polypeptide of claim 41 which comprises amino acids 1-41 of p53.
43. The polypeptide of claim 41 which comprises amino acids 13-57 of p53.
44. The polypeptide of claim 41 which comprises amino acids 1-50 of p53.
45. The preparation of claim 21 wherein the antibodies do not bind to other human proteins.
46. The preparation of claim 21 wherein the antibodies do not bind to human proteins of Mr 75-85K, 105-120K, and 170-200K.
47. The preparation of claim 21 wherein the antibodies bind to the epitope bound by antibodies secreted by hybridoma IF2 (ATCC HB 11290).
48. The preparation of claim 21 wherein the antibodies bind to the epitope bound by antibodies secreted by hybridoma ED9 (ATCC HB 11291).
49. The method of claim 7 wherein the antibodies bind to the epitope on hMDM2 bound by antibodies secreted by hybridoma IF2 (ATCC HB 11290).
50. The method of claim 4 wherein human MDM2 protein is detected by immunohistochemistry.
51. The method of claim 50 wherein antibodies are employed in the immunohistochemistry which bind to an epitope on hMDM2 bound by the antibodies secreted by ED9 (ATCC HB 11291).
52. The method of claim 50 wherein antibodies are employed in the immunohistochemistry which bind to an epitope on hMDM2 bound by the antibodies secreted by IF2 (ATCC HB 11290).
53. The method of claim 4 wherein human MDM2 protein is detected by immunoprecipitation.
54. A hybridoma cell having the identifying characteristics of ED9 (ATCC HB 11291).
55. A hybridoma cell having the identifying characteristics of IF2 (ATCC HB 11290).
Description:
FIELD OF THE INVENTION
[0002]The invention relates to the area of cancer diagnostics and therapeutics. More particularly, the invention relates to the detection of a gene which is amplified in certain human tumors.
BACKGROUND OF THE INVENTION
[0003]According to the Knudson model for tumorigenesis (Cancer Research, 1985, vol. 45, p. 1482), there are tumor suppressor genes in all normal cells which, when they become non-functional due to mutation, cause neoplastic development. Evidence for this model has been found in cases of retinoblastoma and colorectal tumors. The implicated suppressor genes in these tumors, RB and p53 respectively, were found to be deleted or altered in many of the tumors studied.
[0004]The p53 gene product, therefore, appears to be a member of a group of proteins which regulate normal cellular proliferation and suppression of cellular transformation. Mutations in the p53 gene have been linked to tumorigenesis, suggesting that alterations in p53 protein function are involved in cellular transformation. The inactivation of the p53 gene has been implicated in the genesis or progression of a wide variety of carcinomas (Nigro et al., 1989, Nature 342:705-708), including human colorectal carcinoma (Baker et al., 1989, Science 244:217-221), human lung cancer (Takahashi et al., 1989, Science 246:491-494; Iggo et al., 1990, Lancet 335:675-679), chronic myelogenous leukemia (Kelman et al, 1989, Proc. Natl. Acad. Sci. USA 86:6783-6787) and osteogenic sarcomas (Masuda et al., 1987, Proc. Natl. Acad. Sci. USA 84:7716-7719).
[0005]While there exists an enormous body of evidence linking p53 gene mutations to human tumorigenesis (Hollstein et al., 1991, Science 253:49-53) little is known about cellular regulators and mediators of p53 function.
[0006]Hinds et al. (Cell Growth & Differentiation, 1:571-580, 1990), found that p53 cDNA clones, containing a point mutation at amino acid residue 143, 175, 273 or 281, cooperated with the activated ras oncogene to transform primary rat embryo fibroblasts in culture. These mutant p53 genes are representative of the majority of mutations found in human cancer. Hollstein et al., 1991, Science 253:49-53. The transformed fibroblasts were found to produce elevated levels of human p53 protein having extended half-lives (1.5 to 7 hours) as compared to the normal (wild-type) p53 protein (20 to 30 minutes).
[0007]Mutant p53 proteins with mutations at residue 143 or 175 form an oligomeric protein complex with the cellular heat shock protein hsc70. While residue 273 or 281 mutants do not detectably bind hsc70, and are poorer at producing transformed foci than the 175 mutant, complex formation between mutant p53 and hsc70 is not required for p53-mediated transformation. Complex formation does, however, appear to facilitate this function. All cell lines transformed with the mutant p53 genes are tumorigenic in a thymic (nude) mice. In contrast, the wild-type human p53 gene does not possess transforming activity in cooperation with ras. Tuck and Crawford, 1989, Oncogene Res. 4:81-96.
[0008]Hinds et al., supra also expressed human p53 protein in transformed rat cells. When the expressed human p53 was immunoprecipitated with two p53 specific antibodies directed against distinct epitopes of p53, an unidentified Mr 90,000 protein was coimmunoprecipitated. This suggested that the rat Mr 90,000 protein is in a complex with the human p53 protein in the transformed rat cell line.
[0009]As mentioned above, levels of p53 protein are often higher in transformed cells than normal cells. This is due to mutations which increase its metabolic stability (Oven et al., 1981, Mol. Cell. Biol. 1:101-110; Reich et al. (1983), Mol. Cell. Biol. 3:2143-2150). The stabilization of p53 has been associated with complex formation between p53 and viral or cellular proteins. (Linzer and Levine, 1979, Cell 17:43-52; Crawford et al., 1981, Proc. Natl. Acad. Sci. USA 78:41-45; Dippold et al., 1981, Proc. Natl. Acad. Sci. USA 78:1695-1699; Lane and Crawford, 1979, Nature (Lond.) 278:261-263; Hinds et al., 1987, Mol. Cell. Biol. 7:2863-2869; Finlay et al., 1988, Mol. Cell. Biol. 8:531-539; Sarnow et al., 1982, Cell. 28:387-394; Gronostajski et al., 1984, Mol. Cell. Biol. 4:442-448; Pinhasi-Kimhi et al., 1986, Nature (Lond) 320:182-185; Ruscetti and Scolnick, 1983, J. Virol. 46:1022-1026; Pinhasi and Oren, 1984, Mol. Cell. Biol. 4:2180-2186; and Sturzbecher et al., 1987, Oncogene 1:201-211.) For example, p53 protein has been observed to form oligomeric protein complexes with the SV40 large T antigen, the adenovirus type 5 E1B-Mr 55,000 protein, and the human papilloma virus type 16 or 18 E6 product. Linzer and Levine, 1979, Cell 17:43-52; Lane and Crawford, 1979, Nature, 278:261-263; Sarnow et al., 1982, Cell 28:387-394; Werness et al., 1990, Science, 248:76-79. Similarly, complexes have been observed of p105RB (the product of the retinoblastoma susceptibility gene) with T antigen (DeCaprio et al., 1988, Cell 54:275-283), the adenovirus EIA protein (Whyte et al., 1988, Nature 334:124-129) and the E7 protein of human papilloma virus 16 or 18 (Munger et al., 1989, EMBO J. 8:4099-4105). It has been suggested that interactions between these viral proteins and p105RB inactivate a growth-suppressive function of p105RB, mimicking deletions and mutations commonly found in the RB gene in tumor cells. In a similar fashion, oligomeric protein complex formation between these viral proteins and p53 may eliminate or alter the function of p53. Finlay et al., 1989, Cell 57:1083-1093.
[0010]Fakharzadeh et al. (EMBO J. 10:1565-1569, 1991) analyzed amplified DNA sequences present in a tumorigenic mouse cell line (i.e., 3T3DM, a spontaneously transformed derivative of mouse Balb/c cells). Studies were conducted to determine whether any of the amplified genes induced tumorigenicity following introduction of the amplified genes into a nontransformed recipient cell (e.g., mouse NIH3T3 or Rat2 cells). The resulting cell lines were tested for tumorigenicity in nude mice. A gene, designated MDM2, which is amplified more than 50-fold in 3T3DM cells, induced tumorigenicity when overexpressed in NIH3T3 and Rat 2 cells. From the nucleotide and predicted amino acid sequence of mouse MDM2 (mMDM2), Fakharzadeh speculated that this gene encodes a potential DNA binding protein that functions in the modulation of expression of other genes and, when present in excess, interferes with normal constraints on cell growth.
SUMMARY OF THE INVENTION
[0011]It is an object of the invention to provide a method for diagnosing a neoplastic tissue, such as sarcoma, in a human.
[0012]It is another object of the invention to provide a cDNA molecule encoding the sequence of human MDM2.
[0013]Yet another object of the invention is to provide a preparation of human MDM2 protein which is substantially free of other human cellular proteins.
[0014]Still another object of the invention is to provide DNA probes capable of hybridizing with human MDM2 genes or mRNA molecules.
[0015]Another object of the invention is to provide antibodies immunoreactive with human MDM2 protein.
[0016]Still another object of the invention is to provide kits for detecting amplification or elevated expression of human MDM2.
[0017]Yet another object of the invention is to provide methods for identifying compounds which interfere with the binding of human MDM2 to human p53.
[0018]A further object of the invention is to provide a method of treating a neoplastic human cell.
[0019]Yet another object of the invention is to provide methods for inhibiting the growth of tumor cells which contain a human MDM2 gene amplification.
[0020]Still another object of the invention is to provide polypeptides which interfere with the binding of human MDM2 to human p53.
[0021]A further object of the invention is to provide a method for growing host cells containing a p53 expression vector.
[0022]It has now been discovered that hMDM2, a heretofore unknown human gene, plays a role in human cancer. The hMDM2 gene has been cloned and the recombinant derived hMDM2 protein shown to bind to human p53 in vitro. hMDM2 has been found to be amplified in some neoplastic cells and the expression of hMDM2-encoded products has been found to be correspondingly elevated in tumors with amplification of this gene. The elevated levels of MDM2 appear to sequester p53 and allow the cell to escape from p53-regulated growth.
BRIEF DESCRIPTION OF THE DRAWINGS
[0023]FIGS. 1A(1), (2), and (3), 1B(1), (2), and (3), and 1C(1), (2), and (3) show the cDNA sequence of human MDM2. In this figure, human (SEQ ID NO: 2) and mouse (SEQ ID NO: 4) nucleotide and amino acid sequences (SEQ ID NO: 3 and SEQ ID NO: 5, respectively) are compared, the mouse sequence being shown only where it differs from the corresponding human sequence.
[0024]FIG. 2 shows that hMDM2 binds to p53.
[0025]FIG. 3 illustrates the amplification of the hMDM2 gene in sarcomas.
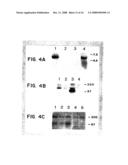
[0026]FIG. 4A-C illustrates hMDM2 expression.
[0027]FIG. 5 shows the inhibition of p53-mediated transactivation by MDM2. Yeast were stably transfected with expression plasmids encoding p53, lex-VP16, MDM2 or the appropriate vector-only controls, as indicated. p53-responsive (bars a-c) or lexA-responsive (bars d-f) β-galactosidase reporter plasmids were used to assess the response. Inset: Western blot analysis demonstrating MDM2 (90 kD) and p53 (53 kD) expression in representative yeast strains. The strain indicated by a plus was transfected with expression vector encoding full length MDM2 and p53, while the strain indicated by a minus was transfected only with the p53 expression vector.
[0028]FIG. 6 shows the determination of MDM2 and p53 domains of interaction. FIG. 5A and FIG. 5B. Random fragments of MDM2 were fused to sequences encoding the lexA DNA binding domain and the resultant clones transfected into yeast carrying pRS314SN (p53 expression vector) and pJK103 (lexA-responsive β-galactosidase reporter). Yeast clones expressing β-galactosidase were identified by their blue color, and the MDM2 sequences in the lexA fusion vector were determined. β-galactosidase activity was observed independent of p53 expression in A, but was dependent on p53 expression in B. The bottom 6 clones in B were generated by genetic engineering. FIG. 6C. Random fragments of p53 were fused to the sequence encoding the B42 acidic activation domain and a hemagglutinin epitope tag; the resultant clones were transfected into yeast carrying lexA-MDM2 (lexA DNA binding domain fused to full length MDM2) and pJK103. Yeast clones were identified as above, and all were found to be MDM2-dependent. The bottom three clones were generated by genetic engineering.
[0029]FIGS. 7A and 7B show protein expression from the yeast strains described in FIG. 6. Western blot analysis was performed as described (Oliner, J. D., et al., Nature 358:80-83 (1992)), using 20 μg of protein per lane. The MDM2 and p53 codons contained in the fusion vectors are shown at the top of A and B, respectively. FIG. 7A. Upper panel probed with p53 Ab2 detecting p53; lower panel probed with anti-lexA polyclonal antibodies (lex Ab) detecting MDM2 fusion proteins of 30-50 kD. FIG. 7B. Upper panel probed with Lex Ab detecting the lexA-full length MDM2 fusion protein of 112 kD; lower panel probed with HA Ab (a monoclonal antibody directed against the hemagglutinin epitope tag, Berkeley Antibody) detecting p53 fusion proteins of approximately 25-30 kD.
[0030]FIG. 8 shows the inhibition of the p53 activation domain by MDM2. Yeast were transfected with expression vectors encoding a lexA-p53 (p53 codons 1-73) fusion (bars a and b) or lexA alone (bar c). Strain b also expressed full length MDM2, and all strains contained the lexA-responsive β-galactosidase reporter plasmid. Inset: Upper panel probed with MDM2 polyclonal antibodies detecting full length MDM2 (90 kD); lower panel probed with lex Ab detecting the lex-p53 fusion protein of 40 kD.
DETAILED DESCRIPTION OF THE INVENTION
[0031]It is a discovery of the present invention that a gene exists which is amplified in some human tumors. The amplification of this gene, designated MDM2, is diagnostic of neoplasia or the potential therefor. Detecting the elevated expression of human MDM2-encoded products is also diagnostic of neoplasia or the potential for neoplastic transformation. Over a third of the sarcomas surveyed, including the most common bone and soft tissue forms, were found to have amplified hMDM2 sequences. Expression of hMDM2 was found to be correspondingly elevated in tumors with the gene amplification.
[0032]Other genetic alterations leading to elevated hMDM2 expression may be involved in tumorigenesis also, such as mutations in regulatory regions of the gene. Elevated expression of hMDM2 may also be involved in tumors other than sarcomas including but not limited to those in which p53 inactivation has been implicated. These include colorectal carcinoma, lung cancer and chronic myelogenous leukemia.
[0033]According to one embodiment of the invention, a method of diagnosing a neoplastic tissue in a human is provided. Tissue or body fluid is isolated from a human, and the copy number of human MDM2 genes is determined. Alternatively, expression levels of human MDM2 gene products can be determined. These include protein and mRNA.
[0034]Body fluids which may be tested include urine, serum, blood, feces, saliva, and the like. Tissues suspected of being neoplastic are desirably separated from normal appearing tissue for analysis. This can be done by paraffin or cryostat sectioning or flow cytometry, as is known in the art. Failure to separate neoplastic from non-neoplastic cells can confound the analysis. Adjacent non-neoplastic tissue or any normal tissue can be used to determine a base-line level of expression or copy number, against which the amount of hMDM2 gene or gene products can be compared.
[0035]The human MDM2 gene is considered to be amplified if the cell contains more than the normal copy number (2) of this gene per genome. The various techniques for detecting gene amplification are well known in the art. Gene amplification can be determined, for example, by Southern blot analysis, as described in Example 4, wherein cellular DNA from a human tissue is digested, separated, and transferred to a filter where it is hybridized with a probe containing complementary nucleic acids. Alternatively, quantitative polymerase chain reaction (PCR) employing primers can be used to determine gene amplification. Appropriate primers will bind to sequences that bracket human MDM2 coding sequences. Other techniques for determining gene copy number as are known in the art can be used without limitation.
[0036]The gene product which is measured may be either mRNA or protein. The term elevated expression means an increase in mRNA production or protein production over that which is normally produced by non-cancerous cells. Although amplification has been observed in human sarcomas, other genetic alterations leading to elevated expression of MDM2 may be present in these or other tumors. Other tumors include those of lung, breast, brain, colorectal, bladder, prostate, liver, skin, and stomach. These, too, are contemplated by the present invention. Non-cancerous cells for use in determining base-line expression levels can be obtained from cells surrounding a tumor, from other humans or from human cell lines. Any increase can have diagnostic value, but generally the mRNA or protein expression will be elevated at least about 3-fold, 5-fold, and in some cases up to about 100-fold over that found in non-cancerous cells. The particular technique employed for detecting mRNA or protein is not critical to the practice of the invention. Increased production of mRNA or protein may be detected, for example, using the techniques of Northern blot analysis or Western blot analysis, respectively, as described in Example 4 or other known techniques such as ELISA, immunoprecipitation, RIA and the like. These techniques are also well known to the skilled artisan.
[0037]According to another embodiment of the invention, nucleic acid probes or primers for the determining of human MDM2 gene amplification or elevated expression of mRNA are provided. The probe may comprise ribo- or deoxyribonucleic acids and may contain the entire human MDM2 coding sequence, a sequence complementary thereto, or fragments thereof. A probe may contain, for example, nucleotides 1-949, or 1-2372 as shown in FIG. 1. Generally, probes or primers will contain at least about 14 contiguous nucleotides of the human sequence but may desirably contain about 40, 50 or 100 nucleotides. Probes are typically labelled with a fluorescent tag, a radioisotope, or the like to render them easily detectable. Preferably the probes will hybridize under stringent hybridization conditions. Under such conditions they will not hybridize to mouse MDM2. The probes of the invention are complementary to the human MDM2 gene. This means that they share 100% identity with the human sequence.
[0038]hMDM2 protein can be produced, according to the invention, substantially free of other human proteins. Provided with the DNA sequence, those of skill in the art can express the cDNA in a non-human cell. Lysates of such cells provide proteins substantially free of other human proteins. The lysates can be further purified, for example, by immunoprecipitation, co-precipitation with p53, or by affinity chromatography.
[0039]The antibodies of the invention are specifically reactive with hMDM2 protein. Preferably, they do not cross-react with MDM2 from other species. They can be polyclonal or monoclonal, and can be raised against native hMDM2 or a hMDM2 fusion protein or synthetic peptide. The antibodies are specifically immunoreactive with hMDM2 epitopes which are not present on other human proteins. Some antibodies are reactive with epitopes unique to human MDM2 and not present on the mouse homolog. The antibodies are useful in conventional analyses, such as Western blot analysis, ELISA, immunohistochemistry, and other immunological assays for the detection of proteins. Techniques for raising and purifying polyclonal antibodies are well known in the art, as are techniques for preparing monoclonal antibodies. Antibody binding can be determined by methods known in the art, such as use of an enzyme-labelled secondary antibody, staphylococcal protein A, and the like. Certain monoclonal antibodies of the invention have been deposited at the American Type Culture Collection, 12301 Parklawn Drive, Rockville, Md. 20852. These include IF2, and ED9, which have been granted accession nos. HB 111290, and HB 11291, respectively.
[0040]According to another embodiment of the invention, interference with the expression of MDM2 provides a therapeutic modality. The method can be applied in vivo, in vitro, or ex vivo. For example, expression may be down-regulated by administering triple-strand forming or antisense oligonucleotides which bind to the hMDM2 gene or mRNA, respectively, and prevent transcription or translation. The oligonucleotides may interact with unprocessed pre-mRNA or processed mRNA. Small molecules and peptides which specifically inhibit MDM2 expression can also be used. Similarly, such molecules which inhibit the binding of MDM2 to p53 would be therapeutic by alleviating the sequestration of p53.
[0041]Such inhibitory molecules can be identified by screening for interference of the hMDM2/p53 interaction where one of the binding partners is bound to a solid support and the other partner is labeled. Antibodies specific for epitopes on hMDM2 or p53 which are involved in the binding interaction will interfere with such binding. Solid supports which may be used include any polymers which are known to bind proteins. The support may be in the form of a filter, column packing matrix, beads, and the like. Labeling of proteins can be accomplished according to any technique known in the art. Radiolabels, enzymatic labels, and fluorescent labels can be used advantageously. Alternatively, both hMDM2 and p53 may be in solution and bound molecules separated from unbound subsequently. Any separation technique known in the art may be employed, including immunoprecipitation or immunoaffinity separation with an antibody specific for the unlabeled binding partner.
[0042]It has been found that amino acid residues 13-41 of p53 (See SEQ ID NO:1) are necessary for the interaction of MDM-2 and p53. However, additional residues on either the amino or carboxy terminal side of the peptide appear also to be required. Nine to 13 additional p53 residues are sufficient to achieve MDM2 binding, although less may be necessary. Since cells which overexpress MDM2 escape from p53-regulated growth control in sarcomas, the use of p53-derived peptides to bind to excess MDM2 leads to reestablishment of p53-regulated growth control.
[0043]Suitable p53-derived peptides for administration are those which are circular, linear, or derivitized to achieve better penetration of membranes, for example. Other organic compounds which are modelled to achieve the same three dimensional structure as the peptide of the invention can also be used.
[0044]DNA encoding the MDM2-binding, p53-derived peptide, or multiple copies thereof, may also be administered to tumor cells as a mode of administering the peptide. The DNA will typically be in an expression construct, such as a retrovirus, DNA virus, or plasmid vector, which has the DNA elements necessary for expression properly positioned to achieve expression of the MDM2-binding peptide. The DNA can be administered, inter alia encapsulated in liposomes, or in any other form known to the art to achieve efficient uptake by cells. As in the direct administration of peptide, the goal is to alleviate the sequestration of p53 by MDM2.
[0045]A cDNA molecule containing the coding sequence of hMDM2 can be used to produce probes and primers. In addition, it can be expressed in cultured cells, such as E. coli, to yield preparations of hMDM2 protein substantially free of other human proteins. The proteins produced can be purified, for example, with immunoaffinity techniques using the antibodies described above.
[0046]Kits are provided which contain the necessary reagents for determining gene copy number, such as probes or primers specific for the hMDM2 gene, as well as written instructions. The instructions can provide calibration curves to compare with the determined values. Kits are also provided to determine elevated expression of mRNA (i.e., containing probes) or hMDM2 protein (i.e., containing antibodies). Instructions will allow the tester to determine whether the expression levels are elevated. Reaction vessels and auxiliary reagents such as chromogens, buffers, enzymes, etc. may also be included in the kits.
[0047]The human MDM2 gene has now been identified and cloned. Recombinant derived hMDM2 has been shown to bind to human p53. Moreover, it has been found that hMDM2 is amplified in some sarcomas. The amplification leads to a corresponding increase in MDM2 gene products. Such amplification is associated with the process of tumorigenesis. This discovery allows specific assays to be performed to assess the neoplastic or potential neoplastic status of a particular tissue.
[0048]The following examples are provided to exemplify various aspects of the invention and are not intended to limit the scope of the invention.
EXAMPLES
Example 1
[0049]To obtain human cDNA clones, a cDNA library was screened with a murine MDM2 (mMDM2) cDNA probe. A cDNA library was prepared by using polyadenylated RNA isolated from the human colonic carcinoma cell line CaCo-2 as a template for the production of random hexamer primed double stranded cDNA. Gubler and Hoffmann, 1983, Gene 25:263-268. The cDNA was ligated to adaptors and then to the lambda YES phage vector, packaged, and plated as described by Elledge et al. (Proc. Natl. Acad. Sci. USA, 88:1731-1735, 1991). The library was screened initially with a P-labelled (Kinzler, K. W., et al., Nucl. Acids Res. 17:3645-3653 (1989), Feinberg and Vogelstein, 1983, Anal. Biochem. 132:6-13) mMDM2 cDNA probe (nucleotides 259 to 1508 (Fakharzadeh et al., 1991, EMBO J. 10:1565-1569)) and then rescreened with an hMDM2 cDNA clone containing nucleotides 40 to 702.
[0050]Twelve clones were obtained, and one of the clones was used to obtain thirteen additional clones by re-screening the same library. In total, twenty-five clones were obtained, partially or totally sequenced, and mapped. Sequence analysis of the twenty-five clones revealed several cDNA forms indicative of alternative splicing. The sequence shown in FIG. 1 is representative of the most abundant class and was assembled from three clones: c14-2 (nucleotides 1-949), c89 (nucleotides 467-1737), and c33 (nucleotides 390-2372). The 3' end of the untranslated region has not yet been cloned in mouse or human. The 5' end is likely to be at or near nucleotide 1. There was an open reading frame extending from the 5' end of the human cDNA sequence to nucleotide 1784. Although the signal for translation initiation could not be unambiguously defined, the ATG at nucleotide 312 was considered the most likely position for several reasons. First, the sequence similarity between hMDM2 and mMDM2 fell off dramatically upstream of nucleotide 312. This lack of conservation in an otherwise highly conserved protein suggested that the sequences upstream of the divergence may not code for protein. Second, an anchored polymerase chain reaction (PCR) approach was employed in an effort to acquire additional upstream cDNA sequence. Ochman et al., 1985, In: PCR Technology: Principles and Applications for DNA Amplification (Erlich, ed.) pp. 105-111 (Stockton, N.Y.). The 5' ends of the PCR derived clones were very similar (within 3 bp) to the 5' ends of clones obtained from the cDNA library, suggesting that the 5' end of the hMDM2 sequence shown in FIG. 1 may represent the 5' end of the transcript. Third, in vitro translation of the sequence shown in FIG. 1, beginning with the methionine encoded by the nucleotide 312 ATG, generated a protein similar in size to that observed in human cells.
[0051]In FIG. 1, hMDM2 cDNA sequence, hMDM2 and mMDM2 nucleotide and amino acid sequences are compared. The mouse sequence is only shown where it differs from the corresponding human sequence. Asterisks mark the 5' and 3' boundaries of the previously published mMDM2 cDNA. Fakharzadeh et al., 1991, EMBO J. 10:1565-1569. Dashes indicate insertions. The mouse and human amino acid sequences are compared from the putative translation start site at nucleotide 312 through the conserved stop codon at nucleotide 1784.
[0052]Comparison of the human and mouse MDM2 coding regions revealed significant conservation at the nucleotide (80.3%) and amino acid (80.4%) levels. Although hMDM2 and mMDM2 bore little similarity to other genes recorded in current databases, the two proteins shared several motifs. These included a basic nuclear localization signal (Tanaka, 1990, FEBS Letters 271:41-46) at codons 181 to 185, several casein kinase II serine phosphorylation sites (Pinna, 1990, Biochem. et. Biophys. Acta. 1054:267-284) at codons 166 to 169, 192 to 195, 269 to 272, and 290 to 293, an acidic activation domain (Ptashne, 1988, Nature 355:683-689) at codons 223 to 274, and two metal binding sites (Harrison, 1991, Nature 353:715) at codons 305 to 322 and 461 to 478, neither of which is highly related to known DNA binding domains. The protein kinase A domain noted in mMDM2 (Fakharzadeh et al., 1991, EMBO J. 10:1565-1569) was not conserved in hMDM2.
Example 2
[0053]To determine whether the hMDM2 protein could bind to human p53 protein in vitro, an hMDM2 expression vector was constructed from the cDNA clones. The hMDM2 expression vector was constructed in pBluescript SK+ (Stratagene) from overlapping cDNA clones. The construct contained the sequence shown in FIG. 1 from nucleotide 312 to 2176. A 42 bp black bettle virus ribosome entry sequence (Dasmahapatra et al., 1987, Nucleic Acid Research 15:3933) was placed immediately upstream of this hMDM2 sequence in order to obtain a high level of expression. This construct, as well as p53 (El-Deriy et al., 1992, Nature Genetics, in press) and MCC (Kinzler et al., 1991, Science 251:1366-1370) constructs in pBluescript SK+, were transcribed with T7 RNA polymerase and translated in a rabbit reticulocyte lysate (Promega) according to the manufacturer's instructions.
[0054]Although the predicted size of the protein generated from the construct was only 55.2 kd (extending from the methionine at nucleotide 312 to nucleotide 1784), in vitro translated protein migrated at approximately 95 kilodaltons.
[0055]Ten μl of lysate containing the three proteins (hMDM2, p53 and MCC), alone or mixed in pairs, were incubated at 37° C. for 15 minutes. One microgram (10 μl) of p53 Ab1 (monoclonal antibody specific for the C-terminus of p53) or Ab2 (monoclonal antibody specific for the N-terminus of p53) (Oncogene Science), or 5 μl of rabbit serum containing MDM2 Ab (polyclonal rabbit anti-hMDM2 antibodies) or preimmune rabbit serum (obtained from the rabbit which produced the hMDM2 Ab), were added as indicated. The polyclonal rabbit antibodies were raised against an E. coli-produced hMDM2-glutathione S-transferase fusion protein containing nucleotides 390 to 816 of the hMDM2 cDNA. Ninety μl of RIPA buffer (10 mM tris [pH 7.5], 1% sodium deoxycholate, 1% NP40, 150 mM NaCl, 0.1% SDS), SNNTE buffer, or Binding Buffer (El-Deriy et al., 1992, Nature Genetics, in press) were then added and the mixtures allowed to incubate at 4° C. for 2 hours.
[0056]Two milligrams of protein A sepharose were added to each tube, and the tubes were rotated end-over-end at 4° C. for 1 hour. After pelleting and washing, the immunoprecipitates were subjected to SDS-polyacrylamide gel electrophoresis and the dried gels autoradiographed for 10 to 60 minutes in the presence of Enhance (New England Nuclear).
[0057]FIG. 2 shows the co-precipitation of hMDM2 and p53. The three buffers produced similar results, although the co-precipitation was less efficient in SNNTE buffer containing 0.5 M NaCl (FIG. 2, lanes 5 and 8) than in Binding Buffer containing 0.1 M NaCl (FIG. 2 lanes 6 and 9).
[0058]In vitro translated hMDM2, p53 and MCC proteins were mixed as indicated above and incubated with p53 Ab1, p53 Ab2, hMDM2 Ab, or preimmune serum. Lanes 1, 4, 7, 10 and 14 contain aliquots of the protein mixtures used for immunoprecipitation. The bands running slightly faster than p53 are polypeptides produced from internal translation initiation sites.
[0059]The hMDM2 protein was not immunoprecipitated with monoclonal antibodies to either the C-terminal or N-terminal regions of p53 (FIG. 2, lanes 2 and 3). However, when in vitro translated human p53 was mixed with the hMDM2 translation product, the anti-p53 antibodies precipitated hMDM2 protein along with p53, demonstrating an association in vitro (FIG. 2, lanes 5 and 6). As a control, a protein of similar electrophoretic mobility from another gene (MCC (Kinzler et al., 1991, Science 251:1366-1370)) was mixed with p53. No co-precipitation of the MCC protein was observed (FIG. 2, lanes 8 and 9). When an in vitro translated mutant form of p53 (175his) was mixed with hMDM2 protein, a similar co-precipitation of hMDM2 and p53 proteins was also observed.
[0060]In the converse of the experiments described above, the anti-hMDM2 antibodies immunoprecipitated p53 when mixed with hMDM2 protein (FIG. 2, lane 15) but failed to precipitate p53 alone (FIG. 5, lane 13). Preimmune rabbit serum failed to precipitate either hMDM2 or p53 (FIG. 2, lane 16).
Example 3
[0061]In order to ascertain the chromosomal localization of hMDM2, somatic cell hybrids were screened with an hMDM2 cDNA probe. A human-hamster hybrid containing only human chromosome 12 was found to hybridize to the probe. Screening of hybrids containing portions of chromosome 12 (Turc-Carel et al., 1986, Cancer Genet. Cytogenet. 23:291-299) with the same probe narrowed the localization to chromosome 12q12-14.
Example 4
[0062]Previous studies have shown that this region of chromosome 12 is often aberrant in human sarcomas. Mandahl et al., 1987, Genes Chromosomes & Cancer 1:9-14; Turc-Carel et al., 1986, Cancer Genet. Cytogenet. 23:291-299; Meltzer et al., 1991, Cell Growth & Differentiation 2:495-501. To evaluate the possibility that hMDM2 was genetically altered in such cancers, Southern blot analysis was performed.
[0063]FIG. 3 shows examples of the amplification of the hMDM2 gene in sarcomas. Cellular DNA (5 μg) was digested with EcoRI, separated by agarose gel electrophoresis, and transferred to nylon as described by Reed and Mann (Nucl. Acids Res., 1985, 13:7207-7215). The cellular DNA was derived from five primary sarcomas (lanes 1-4, 6) and one sarcoma cell line (OsA-Cl, lane 5). The filters were then hybridized with an hMDM2 cDNA fragment probe nucleotide 1-949 (see FIG. 1), or to a control probe which identifies fragments of similar size (DCC gene, 1.65 cDNA fragment). Fearon, 1989, Science 247:49-56. Hybridization was performed as described by Vogelstein et al. (Cancer Research, 1987, 47:4806-4813). A striking amplification of hMDM2 sequences was observed in several of these tumors. (See FIG. 3, lanes 2, 3 and 5). Of 47 sarcomas analyzed, 17 exhibited hMDM2 amplification ranging from 5 to 50 fold. These tumors included 7 to 13 liposarcomas, 7 of 22 malignant fibrous histiocytomas (MFH), 3 of 11 osteosarcomas, and 0 and 1 rhabdomyosarcomas. Five benign soft tissue tumors (lipomas) and twenty-seven carcinomas (colorectal or gastric) were also tested by Southern blot analysis and no amplification was observed.
Example 5
[0064]This example illustrates that gene amplification is associated with increased expression.
[0065]FIG. 4A illustrates hMDM2 expression as demonstrated by Northern blot analysis. Because of RNA degradation in the primary sarcomas, only the cell lines could be productively analyzed by Northern blot. RNA was separated by electrophoresis in a MOPS-formaldehyde gel and electrophoretically transferred to nylon filters. Transfer and hybridization were performed as described by Kinzler et al. (Nature 332:371-374, 1988). The RNA was hybridized to the hMDM2 fragment described in FIG. 3. Ten μg of total RNA derived, respectively, from two sarcoma cell lines (OsA-CL, lane 1 and RC13, lane 2) and the colorectal cancer cell line (CaCo-2) used to make the cDNA library (lane 3). Lane 4 contains 10 μg of polyadenylated CaCo-2 RNA. RNA sizes are shown in kb. In the one available sarcoma cell line with hMDM2 amplification, a single transcript of approximately 5.5 kb was observed (FIG. 4A, lane 1). The amount of this transcript was much higher than in a sarcoma cell line without amplification (FIG. 4A, lane 2) or in a carcinoma cell line (FIG. 4A, lane 3). When purified mRNA (rather than total RNA) from the carcinoma cell line was used for analysis, an hMDM2 transcript of 5.5 kb could also be observed (FIG. 4A, lane 4).
[0066]FIG. 4B illustrates hMDM2 expression as demonstrated by Western blot analysis of the sarcoma cell lines RC13 (lane 1), OsA-CL (lane 3), HOS (lane 4), and the carcinoma cell line CaCo-2 (lane 2).
[0067]FIG. 4C illustrates hMDM2 expression as demonstrated by Western blot analysis of primary sarcomas. Lanes 1 to 3 contain protein from sarcomas with hMDM2 amplifications, and lanes 4 and 5 contain protein from sarcomas without hMDM2 amplification.
[0068]Western blots using affinity purified MDM2 Ab were performed with 50 μg protein per lane as described by Kinzler et al. (Mol. Cell. Biol., 1990, 10:634-642), except that the membranes were blocked in 10% nonfat dried milk and 10% goat serum, and secondary antibodies were coupled to horseradish peroxidase, permitting chemiluminescent detection (Amersham ECL). MDM2 Ab was affinity purified with a pATH-hMDM2 fusion protein using methods described in Kinzler et al. (Mol. Cell. Biol. 10:634-642, 1990). Non-specifically reactive proteins of about 75-85, 105-120 and 170-200 kd were observed in all lanes, irrespective of hMDM2 amplification status. hMDM2 proteins, of about 90-97 kd, were observed only in the hMDM2-amplified tumors. Protein marker sizes are shown in kd.
[0069]A protein of approximately 97 kilodaltons was expressed at high levels in the sarcoma cell line with hMDM2 amplification (FIG. 4B, lane 3), whereas no expression was evident in two sarcoma cell lines without amplification or in the carcinoma cell line (FIG. 4B, lanes 1, 2 and 4). Five primary sarcomas were also examined by Western blot analysis. Three primary sarcomas with amplification expressed the same size protein as that observed in the sarcoma cell line (FIG. 4C, lanes 1-3), while no protein was observed in the two sarcomas without amplification (FIG. 4C, lanes 4 and 5).
[0070]Expression of the hMDM2 RNA in the sarcoma with amplification was estimated to be at least 30 fold higher than that in the other lines examined. This was consistent with the results of Western blot analysis.
[0071]The above examples demonstrate that hMDM2 binds to p53 in vitro and is genetically altered (i.e., amplified) in a significant fraction of sarcomas, including MFH, liposarcomas, and osteosarcomas. These are the most common sarcomas of soft tissue and bone. Weiss and Enzinger, 1978, Cancer 41:2250-2266; Malawer et al., 1985, In: Cancer: Principles and Practice of Oncology, DeVita et al., Eds., pp. 1293-1342 (Lippincott, Philadelphia).
[0072]Human MDM2 amplification is useful for understanding the pathogenesis of these often lethal cancers.
[0073]MDM2 may functionally inactivate p53 in ways similar to those employed by virally encoded oncoproteins such as SV40 T-antigen, adenovirus E1B, and HPV E6. Lane and Bechimol, 1990, Genes and Development 4:1-8; Werness et al., 1990, Science 248:76. Consistent with this hypothesis, no sarcomas with hMDM2 amplification had any of the p53 gene mutations that occur commonly in other tumors. hMDM2 amplification provides a parallel between viral carcinogenesis and the naturally occurring genetic alterations underlying sporadic human cancer. The finding that expression of hMDM2 is correspondingly elevated in tumors with amplification of the gene are consistent with the finding that MDM2 binds to p53, and with the hypothesis that overexpression of MDM2 in sarcomas allows escape from p53 regulated growth control. This mechanism of tumorigenesis has striking parallels to that previously observed for virally induced tumors (Lane and Bechimol, 1990, Genes and Development 4:1-8; Werness et al., 1990, Science 248:76), in which viral oncogene products bind to and functionally inactivate p53.
Example 6
[0074]This example demonstrates that MDM2 expression inhibits p53-mediated transactivation.
[0075]To determine if MDM2 could influence the ability of p53 to activate transcription, expression vectors coding for the two proteins were stably transfected into yeast along with a p53-responsive reporter construct. The reporter consisted of a β-galactosidase gene under the transcriptional control of a minimal promoter and a multimerized human DNA sequence which strongly bound p53 in vitro (Kern, S. E., et al., Science 256:827-830 (1992). Reporter expression was completely dependent on p53 in this assay (FIG. 5, compare bars a and c). MDM2 expression was found to inhibit p53-mediated transactivation of this reporter 16-fold relative to isogeneic yeast lacking MDM2 expression (FIG. 5, compare bars a and b). Western blot analysis confirmed that p53 (53 kD) was expressed equivalently in strains with and without MDM2 (90 kD) (FIG. 1, inset). [0076]METHODS. The MDM2 expression plasmid, pPGK-MDM2, was constructed by inserting the full length MDM2 cDNA (Oliner, J. D., et al., Nature 358:80-83 (1992)) into pPGK (Poon, D. et al., Mol. and Cell. Biol. 1111:4809-4821 (1991)), immediately downstream of the phosphoglycerate kinase constitutive promoter. Galactose-inducible p53 (pRS314SN, Nigro, J. M., et al., Mol. and Cell. Biol. 12:1357-1365 (1992)), lexA-VP16 (YVLexA, Dalton, S., et al., Cell 68:597-612 (1992)), and lexA (YLexA, YVLexA minus VP16) plasmids were used as indicated. The reporters were PG16-lacZ (Kern, S. E. et al., Science 256:827-830 (1992)) (p53-responsive) and pJK103 (Kamens, J., et al., Mol. Cell. Biol 10:2840-2847 (1990)) (lexA-responsive). S. cerevisiae strain pEGY48 was transformed as described (Kinzler, K. W. et al., Nucl. Acids Res. 17:3645-3653 (1989)). Yeast strains represented by bars a-c were grown at 30° C. to mid-log phase in selective liquid medium containing 2% raffinose as the carbon source, induced for 30 minutes by the addition of 2% galactose, harvested, and lysed as described (Kern, S. E. et al., Science 256:827-830 (1992)). The strains represented by bars d-f were treated similarly, except that the cells were induced in galactose for 4 hours to obtain measurable levels of β-galactosidase. β-galactosidase activities shown represent the mean of three to five experimental values (error bars indicate s.e.m.). Protein concentrations were determined by a Coomassie blue-based (bio-Rad) assay. The β-galactosidase assays were performed with AMPGD chemiluminescent substrate and Emerald enhancer (Tropix) according to the manufacturer's instructions. β-galactosidase activities of bars b and c are shown relative to that of bar A; β-galactosidase activities of bars e and f are shown relative to that of bar d. Western blots were performed as described (Oliner, J. D., et al., Nature 358:80-83 (1992)), using p53 Ab1801 (lower panel, Oncogene Science) or MDM2 polyclonal antibodies (Oliner, J. D., et al., Nature 358:80-83 (1992)) (upper panel).
[0077]To ensure that this inhibition was not simply a general transcriptional down regulation mediated by the expression of the foreign MDM2 gene, a yeast strain was created that contained a different transcriptional activator (lexA-VP16, consisting of the lexa DNA binding domain fused to the VP16 acidic activation domain), a similar reporter (with a lexA-responsive site upstream of a β-galactosidase gene), and the same MDM2 expression vector. The results shown in FIG. 1 (bars d & e) demonstrate that lexA-VP16 transactivation was unaffected by the presence of MDM2. Furthermore, MDM2 expression had no apparent effect on the growth rate of the cells.
Example 7
[0078]This example demonstrates the domains of p53 and MDM2 which interact with each other.
[0079]To gain insight into the mechanism of the MDM2-mediated p53 inhibition, the domains of MDM2 and p53 responsible for binding to one another were mapped. The yeast system used to detect protein-protein binding takes advantage of the modular nature of transcription factor domains (Keegan, L., et al., Science 231:699-704 (1986); Chien, C.-T., Proc. Natl. Acad. Sci. U.S.A. 88:9578-9582 (1991); Brent, R., et al., Cell 43:729-731 (1985); Ma, J., et al., Cell 55:4430446 (1988). Generically, if protein 1 (fused to a sequence-specific DNA binding domain) is capable of binding to protein 2 (fused to a transcriptional activation domain), then co-expression of both fusion proteins will result in transcriptional activation of a suitable reporter. In our experiments, the lexA DNA binding domain (amino acids 2-202) and the B42 acidic activation domain (AAD) were used in the fusion constructs. The reporter (Kamens, J., et al., Mol. Cell. Biol. 10:2840-2847 (1990); contained a lexA-responsive site upstream of a β-galactosidase gene. As an initial control experiment, full length MDM2 was inserted into the lexA fusion vector, and full length p53, supplying its intrinsic activation domain was inserted into a non-fusion vector. The combination resulted in the activation of the lexA-responsive reporter, while the same expression constructs lacking either the MDM2 or p53 cDNA inserts failed to activate β-galactosidase (Table I, strains 1, 2, and 3). Thus, activation was dependent upon MDM2-p53 binding.
[0080]This assay was then applied to mapping the interaction domains of each protein. Full length cDNA fragments encoding MDM2 or p53 were randomly sheared by sonication, amplified by polymerase chain reaction, size fractionated, cloned into the appropriate fusion vectors and transfected into yeast along with the reporter and the full length version of the other protein. [0081]METHODS. Full length MDM2 cDNA in pBluescript SK+ (Stratagene) was digested with XhoI and BamHI to excise the entire insert. After agarose gel purification, the insert was sheared into random fragments by sonication, polished with the Klenow fragment of DNA polymerase I, ligated to catch linkers, and amplified by the polymerase chain reaction as described (Kinzler, K. W., et al., Nucl. Acids Res. 17:3645-3653 (1989)). The fragments were fractionated on an acrylamide gel into size ranges of 100-400 bp or 400-1000 pb, cloned into lexA(1-202)+PL (Ruden, D. M., et al., Nature 350:250-252 (1991)), and transfected into bacteria (XL-1 Blue, Stratagene).
[0082]At least 10,000 bacterial colonies were scraped off agar plates, and the plasmid DNA was transfected into a strain of pEGY48 containing pRS314N (p53 expression vector) and pJK103 (lexA-responsive β-galactosidase reporter). Approximately 5,000 yeast clones were plated on selective medium containing 2% dextrose, and were replica-plated onto glalctose- and X-gal-containing selective medium. Blue colonies (17) appeared only on the plates containing the larger fragments of MDM2. The 17 isolated colonies were tested for blue color in this assay both in the presence and in the absence of galactose (p53 induction); all tested positive in the presence of galactose but only 2 of the 17 tested positive in its absence. MDM2-containing plasmid DNA extracted from the 17 yeast clones was selectively transferred to bacterial strain KC8 and sequenced from the lexA-MDM2 junction. The MDM2 sequences of the two p53-independent clones are diagrammed in FIG. 6A. The MDM2 sequences of the remaining 15 p53-dependent clones coded for peptides ranging from 135 to 265 a.a. in length and began exclusively at the initiator methionine. Three of the MDM2 sequences obtained are shown at the top of FIG. 6B. The lower 6 sequences were genetically engineered (using the polymerase chain reaction and appropriate primers) into lexA(1-202)+PL and subsequently tested to further narrow the binding region. [0083]Fragments of p53 were also cloned into pJG4-5, producing a fusion protein C-terminal to the B42 acidic activation domain and incorporating an epitope of hemagglutinin. The clones were transfected into a strain of pEGY48 already containing lex-MDM2 (plex-202+PL containing full length MDM2) and pJK103. The top three p53 sequences shown in FIG. 6C. were derived from yeast obtained by colony screening, whereas the lower three were genetically engineered to contain the indicated fragments.
[0084]The resultant yeast colonies were examined for β-galactosidase activity in situ. Of approximately 5000 clones containing MDM2 fragments fused to the lexA DNA binding domain, 17 were found to score positively in this assay. The clones could be placed into two classes. The first class (two clones) expressed low levels of β-galactosidase (about 5-fold less than the other fifteen clones) and β-galactosidase expression was independent of p53 expression (FIG. 6A). These two clones encoded MDM2 amino acids 190-340 and 269-379, respectively. The region shared between these two clones overlapped the only acidic domain in MDM2 (amino acids 230-301). This domain consisted of 37.5% aspartic and glutamic acid residues but no basic amino acids. This acidic domain appears to activate transcription only when isolated from the rest of the MDM2 sequence, because the entire MDM2 protein fused to lexA had no measurable β-galactosidase activity in the same assay (Table I, strain 3). The other class (15 clones) each contained the amino terminal region of MDM2 (FIG. 6B). The β-galactosidase activity of these clones was dependent on p53 co-expression. To narrow down the region of interaction, we generated six additional clones by genetic engineering. The smallest tested region of MDM2 which could functionally interact with full length p53 contained MDM2 codons 1 to 118 (FIG. 6B). The relatively large size of the domain required for interaction was consistent with the fact that when small sonicated fragments of MDM2 were used in the screening assay (200 bp instead of 600 bp average size), no positively scoring clones were obtained.
[0085]In a converse set of experiments, yeast clones containing fragments of p53 fused to the B42 AAD were screened for lexA-responsive reporter expression in the presence of a lexA-MDM2 fusion protein. Sequencing of the 14 clones obtained in the screen revealed that they could be divided into three subsets, one containing amino acids 1-41, a second containing amino acids 13-57, and a third containing amino acids 1-50 (FIG. 2C). The minimal overlap between these three fragments contained codons 13-41. Although this minimal domain was apparently necessary for interaction with MDM2, it was insufficient, as the fragments required 9-12 amino acids on either side of codons 13-41 for activity (FIG. 6C). To further test the idea that the amino terminal region of p53 was required for MDM2 binding, we generated an additional yeast strain expressing the lexA-DNA binding domain fused to p53 codons 74-393) and the B42 acidic activation domain fused to full length MDM2. These strains failed to activate the same lexA-responsive reporter (Table I, strain 8), as expected if the N-terminus of p53 were required for the interaction.
TABLE-US-00001 TABLE I STRAIN p53 MDM2 NUMBER CONSTRUCT CONSTRUCT ACTIVATION 1 p53a Vectorb - 2 p53a lexA˜MDM2b + 3 Vectora lexA˜MDM2b - 4 p53a lexA-MDM2 (1-118)b + 5 Vectora lexA-MDM2 (1-118)b - 6 B42-p53 (1-41)c lexA-MDM2b + 7 B42-p53 (1-41)c Vectorb - 8 lexA-p53 (74-393)b B42-MDM2c - 9 p53 (1-137)a lexA-MDM2b - The MDM2 and p53 proteins expressed in each strain, along with the relevant reporters, are indicated. Numbers in parentheses refer to the MDM2 or p53 amino acids encoded (absence of parentheses indicated full length protein, that is, MDM2 amino acids 1 to 491 or p53 amino acids 1 to 393). The lexA-responsive β-galactosidase reporter plasmid (pJK103, Kamens, J., et al., Mol. Cell. Biol. 10: 2840-2847 (1990)) was present in all strains. apRS314 vector (Nigro, J. M., et al., Mol. and Cell. Biol. 12: 1357-1365 (1992). bplex(1-202)+PL vector, containing lexA DNA binding domain fused to insert (Ruden, D. M., et al., Nature 350: 250-252 (1991). cpJG4-5 vector, containing B42 activation domain fused to insert. d(+) indicates that colonies turned blue following 24 hours of incubation on X-gal-containing selective medium, while (-) indicates that colonies remained white following 72 hours of incubation.
[0086]Sequence analysis showed that all p53 and MDM2 fragments noted in FIG. 6 were ligated in frame and in the correct orientation relative to the B42 and lexA domains, respectively. Additionally, all clones compared in FIG. 6 expressed the relevant proteins at similar levels, as shown by Western blotting (FIG. 7).
[0087]The most striking results of these mapping experiments was that the region of p53 required to bind MDM2 was almost identical to the previously identified acidic activation domain of p53 (amino acids 20-42) (Unger, T., et al., EMBO J. 11:1383-1390 (1992); Miller, C. W., et al., Proc. Am. Assoc. Cancer Res. 33:386 (1992). This suggested that MDM2 inhibits p53-mediated transcriptional activation by "concealing" the activation domain of p53 from the transcriptional machinery. If this were true, the p53 activation domain, in isolation from the rest of the p53 protein, should still be inhibitable by full length MDM2. To test this hypothesis, we produced a hybrid protein containing the p53 activation domain (codons 1-73) fused to the lexA-DNA binding domain. This construct exhibited strong transcriptional activation of a lexA-responsive reporter (FIG. 8), as predicted from previous experiments in which the p53 activation domain was fused to another DNA binding domain (Fields, S., et al., Science 249:1046-1049 (1990); Raycroft, L., et al., Science 249:1049-1051 (1990)). The lexA-p53 DNA construct was stably expressed in yeast along with the full length MDM2 expression vector (or the vector alone). MDM2 expression resulted in a five-fold decrease in reporter activity, demonstrating that MDM2 can specifically inhibit the function of the p53 activation domain regardless of the adjacent protein sequences tethering p53 to DNA (FIG. 8). [0088]METHODS. Strains were grown to mid-log phase in 2% dextrose before induction of p53 expression for 2 hours by the addition of 2% galactose. The lex-p53 construct was identical to lex-VP16 (YVlexA, Dalton, S., et al., Cell 68:597-612 (1992)) except that VP16 sequences were replaced by p53 sequences encoding amino acids 1 to 73.
[0089]The results obtained in the experiments discussed herein raise an interesting paradox. If MDM2 binds to (FIG. 6) and conceals (FIG. 8) the p53 activation domain from the transcriptional machinery, how could the lexA-MDM2-p53 complex activate transcription from the lexA-responsive reporter (Table I, strain 2)? Because the only functional activation domain in the lexA-MDM2-p53 complex of strain 2 is expected to be contributed by p53, one might predict that it would be concealed by binding to MDM2 and thereby fail to activate. A potential resolution of this paradox is afforded by knowledge that p53 exists as a homotetramer (Stenger, J. E., et al., Mol. Carcinogenesis. 5:102-106 (1992); Sturzbecher, H. W. et al., Oncogene 7:1513-1523 (1992). Thus the activation noted in the lexA-MDM2-p53 complex could be due to the presence of four individual activation domains contributed by the p53 tetramer, not all of which were concealed by MDM2. As a direct test of this issue, the domain of p53 required for homo-oligomerization (Stenger, J. E., et al., Mol. Carcinogenesis. 5:102-106 (1992); Sturzbecher, H. W. et al., Oncogene 7:1513-1523 (1992) (the C-terminus) was removed from the p53 expression construct, so that it consisted of only codons 1-137. This truncated p53 polypeptide retained the entire activation domain (as shown in FIG. 8, bar a) and the entire domain required for interaction with MDM2 (Table I, strain 6). Yet, when allowed to interact with lexA-MDM2, no transactivation of the lexA-responsive reporter was observed (Table I, strain 9). Because p53 did not inhibit lexA-MDM2 binding to the lexA reporter (Table I, strain 2), the result of strain 9 is likely to be due to a direct inhibition of the isolated p53 activation domain by MDM2.
Example 8
[0090]This example illustrates the production and characterization of antibodies specific for MDM2 epitopes.
[0091]The antigen preparations used to intraperitoneally immunize female (BALB/c×C57BL/6)F1 mice comprised bacterially expressed, glutathione-column purified glutathione-S-transferase-MDM2 (GST-MDM2) fusion protein. (One preparation was further purified on a polyacrylamide gel and electroeluted.) The fusion protein contains a 16 kD amino terminal portion of human MDM2 protein (amino acids 27 to 168). For immunization, the fusion protein was mixed with Ribi adjuvant (Ribi Immunochem Research, Inc.).
[0092]Two mice were sacrificed and their spleen cells fused to SP2/0s myeloma cells (McKenzie, et al., Oncogene, 4:543-548, 1989). Resulting hybridomas were screened by ELISA on trpE-MDM2 fusion protein-coated microtiter wells. The trpE-MDM2 fusion protein contains the same portion of MDM2 as the GST-MDM2 fusion protein. Antigen was coated at a concentration of 1 μg/ml.
[0093]A second fusion was performed as described except hybridomas were screened on electroeluted, glutathione purified GST-MDM2. Positive hybridomas from both fusions were expanded and single cell subcloned. Subclones were tested by Western Blot for specificity to the 55 kD trpE-MDM2 and the 43 kD GST-MDM2 fusion proteins.
[0094]Two Western Blot positive subclones (1F2 and JG3) were put into mice for ascites generation. The resulting ascites were protein A purified. Both purified monoclonal antibodies tested positive by Western Blot and immunoprecipitation for the 90 kD migrating MDM2 protein present in a human osteosarcoma cell line (OsA-CL), which overexpresses MDM2, and negative in the HOS osteosarcoma, which does not overexpress MDM2.
[0095]ED9 was protein G-purified from ascites and found to be specific in cryostat immunohistochemistry for MDM2 in osteosarcoma cells, as was IF2.
Example 9
[0096]This example demonstrates the expression and detection of MDM2 at the cellular level.
[0097]To evaluate MDM2 expression at the cellular level, we produced monoclonal antibodies against bacterially generated fusion proteins containing residues 27 to 168 of MDM2. (See example 8.) Of several antibodies tested, mAb IF-2 was the most useful, as it detected MDM2 in several assays. For initial testing, we compared proteins derived from OsA-CL, a sarcoma cell line with MDM2 amplification but without p53 mutation (Table II) and proteins from SW480, a colorectal cancer cell line with p53 mutation (Barak et al., EMBO 12:461-468 (1993)) but without MDM2 amplification (data not shown). FIG. 9 shows that the mAb IF-2 detected an intense 90 kd band plus several other bands of lower molecular weight in OsA-CL extracts, and a much less intense 90 kd band in SW480 extracts. We could not distinguish whether the low molecular weight bands in OsA-CL were due to protein degradation or alternative processing of MDM2 transcripts. The more than 20-fold difference in intensity between the signals observed in OsA-CL and SW480 is consistent with the greater than 20-fold difference in MDM2 gene copy number in these two lines. Conversely, the 53 kd signal detected with p53-specific mAb 1801 was much stronger in SW480 than in OsA-CL consistent with the presence of a mutated p53 in SW480 (FIG. 9).
[0098]Cells grown on cover slips were then used to assess the cellular localization of the MDM2 protein. A strong signal, exclusively nuclear, was observed in OsA-CL cells with the IF-2 mAb and a weaker signal, again strictly nuclear, was observed in SW480 (FIG. 10). The nuclear localization of MDM2 is consistent with previous studies of mouse cells (Barak et al., EMBO 12:461-468 (1993)) and the fact that human MDM2 contains a nuclear localization signal at residues 179 to 186. Reactivity with the p53-specific antibody was also confined to the nuclei of these two cell lines (FIG. 10), with the relative intensities consistent with the Western blot results (FIG. 9).
[0099]The IF-2 mAb was then used (at 5 μg/ml) to stain the seven primary sarcomas noted above. The nuclei of two of them (tumors #3 and #10) stained strongly (FIG. 11). Both of these tumors contained MDM2 gene amplification (Table II). In the five tumors without amplification, little or no MDM2 reactivity was observed (example in FIG. 11).
TABLE-US-00002 TABLE II TUMOR TUMOR MDM2 P53 OVER- # ID TYPEa AMPLIFICATIONb ALTERATIONc EXPRESSIONd 1 M-2 MFH ABSENT DELETION/ NONE REARRANGEMENT 2 M-5 MFH ABSENT CGC-CUC MUTATION; p53 Arg(158)-His 3 M-7 MFH PRESENT NONE OBSERVED MDM2 4 M-8 MFH ABSENT DELETION NONE 5 M-14 MFH ABSENT NONE OBSERVED N.T. 6 M-15 MFH ABSENT DELETION N.T. 7 M-16 MFH ABSENT NONE OBSERVED NONE 8 M-17 MFH ABSENT NONE OBSERVED N.T. 9 M-18 MFH ABSENT OVEREXPRESSED p53 10 M-20 MFH PRESENT NONE OBSERVED MDM2 11 L-5 LIPOSARCOMA ABSENT NONE OBSERVED N.T. 12 L-7 LIPOSARCOMA ABSENT AAC-AGC MUTATION; N.T. Asn(239)-Ser 13 L-9 LIPOSARCOMA PRESENT NONE OBSERVED N.T. TUMOR TUMOR MDM2 P53 OVER- # ID TYPEa AMPLIFICATIONb MUTATIONc EXPRESSIONd 14 L-1l LIPOSARCOMA ABSENT NONE OBSERVED N.T. 15 KL5B LIPOSARCOMA ABSENT CAG-UAG MUTATION; N.T. Gln(144)-Stop 16 KL7 LIPOSARCOMA PRESENT NONE OBSERVED N.T. 17 KL10 LIPOSARCOMA ABSENT NONE OBSERVED N.T. 18 KL11 LIPOSARCOMA ABSENT GGT-GAT MUTATION; EXON 5 N.T. SPLICE DONOR SITE 19 KL12 LIPOSARCOMA ABSENT NONE OBSERVED N.T. 20 KL28 LIPOSARCOMA PRESENT NONE OBSERVED N.T. 21 KL3O LIPOSARCOMA PRESENT NONE OBSERVED N.T. 22 S189 LIPOSARCOMA PRESENT NONE OBSERVED N.T. 23 S131B LIPOSARCOMA ABSENT NONE OBSERVED N.T. 24 OSA-CL MFH PRESENT NONE OBSERVED MDM2 aMFH = malignant fibrous histiocytoma bas assessed by Southern blot cas assessed by Southern blot, sequencing of exons 5-8, or immunohistochemical analysis das assessed by immunohistochemical analysis; N.T. = not tested
Sequence CWU
1
User Contributions:
comments("1"); ?> comment_form("1"); ?>Inventors list |
Agents list |
Assignees list |
List by place |
Classification tree browser |
Top 100 Inventors |
Top 100 Agents |
Top 100 Assignees |
Usenet FAQ Index |
Documents |
Other FAQs |
User Contributions:
Comment about this patent or add new information about this topic:
Images included with this patent application:
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |  |
 |
| Similar patent applications: | |
| Date | Title |
|---|---|
| 2012-12-20 | Reprogramming a cell by activation of the endogenous transcription factor network |
| 2012-12-20 | Production of cellulases and hemicellulases in algal biofuel feedstocks |
| 2012-12-20 | Covalent joining of dna strands to rna strands catalyzed by vaccinia topoisomerase |
| 2012-12-06 | Generation of hydrogen from hydrocarbon bearing materials |
| 2012-12-20 | Modified yeast strain and a method for producing squalene using the same |
| New patent applications in this class: | |
| Date | Title |
|---|---|
| 2011-06-30 | Apparatus and method of authenticating product using polynucleotides |
| 2011-06-30 | Cyanine compounds, compositions including these compounds and their use in cell analysis |
| 2011-06-30 | Method for detecting multiple small nucleic acids |
| 2011-06-30 | Solid-phase chelators and electronic biosensors |
| 2011-06-30 | Cell-based screening assay to identify molecules that stimulate ifn-alpha/beta target genes |
| New patent applications from these inventors: | |
| Date | Title |
|---|---|
| 2022-09-01 | Checkpoint blockade and microsatellite instability |
| 2022-08-18 | Rapid aneuploidy detection |
| 2022-08-18 | Checkpoint blockade and microsatellite instability |
| Top Inventors for class "Chemistry: molecular biology and microbiology" | |
| Rank | Inventor's name |
|---|---|
| 1 | Marshall Medoff |
| 2 | Anthony P. Burgard |
| 3 | Mark J. Burk |
| 4 | Robin E. Osterhout |
| 5 | Rangarajan Sampath |